User login
Translating the 2019 AAD-NPF Guidelines of Care for the Management of Psoriasis With Biologics to Clinical Practice
Psoriasis is a systemic immune-mediated disorder characterized by erythematous, scaly, well-demarcated plaques on the skin that affects approximately 3% of the world’s population.1 The disease is moderate to severe for approximately 1 in 6 individuals with psoriasis.2 These patients, particularly those with symptoms that are refractory to topical therapy and/or phototherapy, can benefit from the use of biologic agents, which are monoclonal antibodies and fusion proteins engineered to inhibit the action of cytokines that drive psoriatic inflammation.
In February 2019, the American Academy of Dermatology (AAD) and National Psoriasis Foundation (NPF) released an updated set of guidelines for the use of biologics in treating adult patients with psoriasis.3 The prior guidelines were released in 2008 when just 3 biologics—etanercept, infliximab, and adalimumab—were approved by the US Food and Drug Administration (FDA) for the management of psoriasis. These older recommendations were mostly based on studies of the efficacy and safety of biologics for patients with psoriatic arthritis.4 Over the last 11 years, 8 novel biologics have gained FDA approval, and numerous large phase 2 and phase 3 trials evaluating the risks and benefits of biologics have been conducted. The new guidelines contain considerably more detail and are based on evidence more specific to psoriasis rather than to psoriatic arthritis. Given the large repertoire of biologics available today and the increased amount of published research regarding each one, these guidelines may aid dermatologists in choosing the optimal biologic and managing therapy.
The AAD-NPF recommendations discuss the mechanism of action, efficacy, safety, and adverse events of the 10 biologics that have been FDA approved for the treatment of psoriasis as of March 2019, plus risankizumab, which was pending FDA approval at the time of publication and was later approved in April 2019. They also address dosing regimens, potential to combine biologics with other therapies, and different forms of psoriasis for which each may be effective.3 The purpose of this discussion is to present these guidelines in a condensed form to prescribers of biologic therapies and review the most clinically significant considerations during each step of treatment. Of note, we highlight only treatment of adult patients and do not discuss information relevant to risankizumab, as it was not FDA approved when the AAD-NPF guidelines were released.
Choosing a Biologic
Biologic therapy may be considered for patients with psoriasis that affects more than 3% of the body’s surface and is recalcitrant to localized therapies. There is no particular first-line biologic recommended for all patients with psoriasis; rather, choice of therapy should be individualized to the patient, considering factors such as body parts affected, comorbidities, lifestyle, and drug cost.
All 10 FDA-approved biologics (Table) have been ranked by the AAD and NPF as having grade A evidence for efficacy as monotherapy in the treatment of moderate to severe plaque-type psoriasis. Involvement of difficult-to-treat areas may be considered when choosing a specific therapy. The tumor necrosis factor α (TNF-α) inhibitors etanercept and adalimumab, the IL-17 inhibitor secukinumab, and the IL-23 inhibitor guselkumab have the greatest evidence for efficacy in treatment of nail disease. For scalp involvement, etanercept and guselkumab have the highest-quality evidence, and for palmoplantar disease, adalimumab, secukinumab, and guselkumab are considered the most effective. The TNF-α inhibitors are considered the optimal treatment option for concurrent psoriatic arthritis, though the IL-12/IL-23 inhibitor ustekinumab and the IL-17 inhibitors secukinumab and ixekizumab also have shown grade A evidence of efficacy. Of note, because TNF-α inhibitors received the earliest FDA approval, there is most evidence available for this class. Therapies with lower evidence quality for certain forms of psoriasis may show real-world effectiveness in individual patients, though more trials will be necessary to generate a body of evidence to change these clinical recommendations.
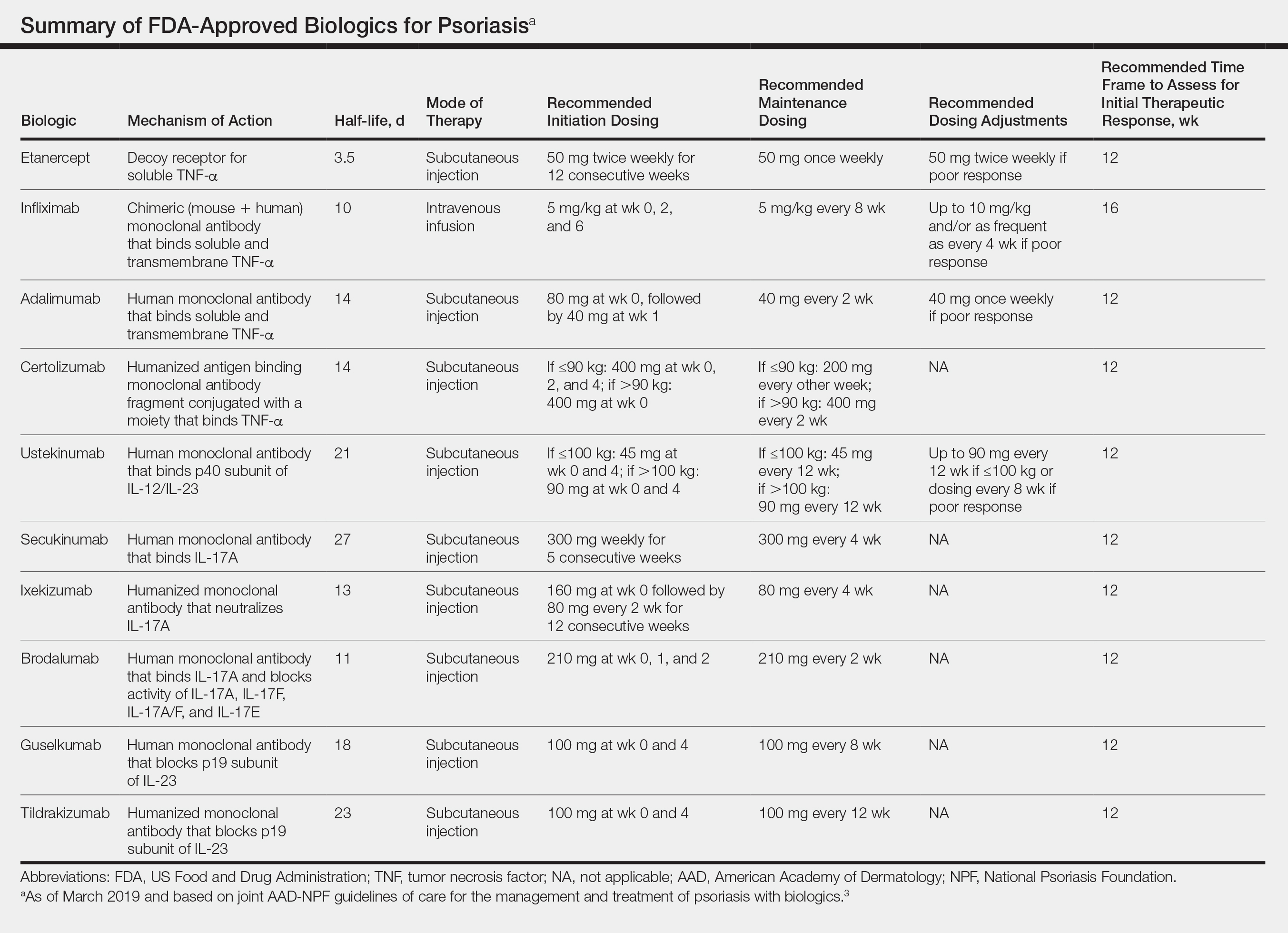
In pregnant women or those are anticipating pregnancy, certolizumab may be considered, as it is the only biologic shown to have minimal to no placental transfer. Other TNF-α inhibitors may undergo active placental transfer, particularly during the latter half of pregnancy,5 and the greatest theoretical risk of transfer occurs in the third trimester. Although these drugs may not directly harm the fetus, they do cause fetal immunosuppression for up to the first 3 months of life. All TNF-α inhibitors are considered safe during lactation. There are inadequate data regarding the safety of other classes of biologics during pregnancy and lactation.
Overweight and obese patients also require unique considerations when choosing a biologic. Infliximab is the only approved psoriasis biologic that utilizes proportional-to-weight dosing and hence may be particularly efficacious in patients with higher body mass. Ustekinumab dosing also takes patient weight into consideration; patients heavier than 100 kg should receive 90-mg doses at initiation and during maintenance compared to 45 mg for patients who weigh 100 kg or less. Other approved biologics also may be utilized in these patients but may require closer monitoring of treatment efficacy.
There are few serious contraindications for specific biologic therapies. Any history of allergic reaction to a particular therapy is an absolute contraindication to its use. In patients for whom IL-17 inhibitor treatment is being considered, inflammatory bowel disease (IBD) should be ruled out given the likelihood that IL-17 could reactivate or worsen IBD. Of note, TNF-α inhibitors and ustekinumab are approved therapies for patients with IBD and may be recommended in patients with comorbid psoriasis. Phase 2 and phase 3 trials have found no reactivation or worsening of IBD in patients with psoriasis who were treated with the IL-23 inhibitor tildrakizumab,6 and phase 2 trials of treatment of IBD with guselkumab are currently underway (ClinicalTrials.gov Identifier NCT03466411). In patients with New York Heart Association class III and class IV congestive heart failure or multiple sclerosis, initiation of TNF-α inhibitors should be avoided. Among 3 phase 3 trials encompassing nearly 3000 patients treated with the IL-17 inhibitor brodalumab, a total of 3 patients died by suicide7,8; hence, the FDA has issued a black box warning cautioning against use of this drug in patients with history of suicidal ideation or recent suicidal behavior. Although a causal relationship between brodalumab and suicide has not been well established,9 a thorough psychiatric history should be obtained in those initiating treatment with brodalumab.
Initiation of Therapy
Prior to initiating biologic therapy, it is important to obtain a complete blood cell count, complete metabolic panel, tuberculosis testing, and hepatitis B virus (HBV) and hepatitis C virus serologies. Testing for human immunodeficiency virus may be pursued at the clinician’s discretion. It is important to address any positive or concerning results prior to starting biologics. In patients with active infections, therapy may be initiated alongside guidance from an infectious disease specialist. Those with a positive purified protein derivative test, T-SPOT test, or QuantiFERON-TB Gold test must be referred for chest radiographs to rule out active tuberculosis. Patients with active HBV infection should receive appropriate referral to initiate antiviral therapy as well as core antibody testing, and those with active hepatitis C virus infection may only receive biologics under the combined discretion of a dermatologist and an appropriate specialist. Patients with human immunodeficiency virus must concurrently receive highly active antiretroviral therapy, show normal CD4+ T-cell count and undetectable viral load, and have no recent history of opportunistic infection.
Therapy should be commenced using specific dosing regimens, which are unique for each biologic (Table). Patients also must be educated on routine follow-up to assess treatment response and tolerability.
Assessment and Optimization of Treatment Response
Patients taking biologics may experience primary treatment failure, defined as lack of response to therapy from initiation. One predisposing factor may be increased body mass; patients who are overweight and obese are less likely to respond to standard regimens of TNF-α inhibitors and 45-mg dosing of ustekinumab. In most cases, however, the cause of primary nonresponse is unpredictable. For patients in whom therapy has failed within the recommended initial time frame (Table), dose escalation or shortening of dosing intervals may be pursued. Recommended dosing adjustments are outlined in the Table. Alternatively, patients may be switched to a different biologic.
If desired effectiveness is not reached with biologic monotherapy, topical corticosteroids, topical vitamin D analogues, or narrowband UVB light therapy may be concurrently used for difficult-to-treat areas. Evidence for safety and effectiveness of systemic adjuncts to biologics is moderate to low, warranting caution with their use. Methotrexate, cyclosporine, and apremilast have synergistic effects with biologics, though they may increase the risk for immunosuppression-related complications. Acitretin, an oral retinoid, likely is the most reasonable systemic adjunct to biologics because of its lack of immunosuppressive properties.
In patients with a suboptimal response to biologics, particularly those taking therapies that require frequent dosing, poor compliance should be considered.10 These patients may be switched to a biologic with less-frequent maintenance dosing (Table). Ustekinumab and tildrakizumab may be the best options for optimizing compliance, as they require dosing only once every 12 weeks after administration of loading doses.
Secondary treatment failure is diminished efficacy of treatment following successful initial response despite no changes in regimen. The best-known factor contributing to secondary nonresponse to biologics is the development of antidrug antibodies (ADAs), a phenomenon known as immunogenicity. The development of efficacy-limiting ADAs has been observed in response to most biologics, though ADAs against etanercept and guselkumab do not limit therapeutic response. Patients taking adalimumab and infliximab have particularly well-documented efficacy-limiting immunogenicity, and those who develop ADAs to infliximab are considered more prone to developing infusion reactions. Methotrexate, which limits antibody formation, may concomitantly be prescribed in patients who experience secondary treatment failure. It should be considered in all patients taking infliximab to increase efficacy and tolerability of therapy.
Considerations During Active Therapy
In addition to monitoring adherence and response to regimens, dermatologists must be heavily involved in counseling patients regarding the risks and adverse effects associated with these therapies. During maintenance therapy with biologics, patients must follow up with the prescriber at minimum every 3 to 6 months to evaluate for continued efficacy of treatment, extent of side effects, and effects of treatment on overall health and quality of life. Given the immunosuppressive effects of biologics, annual testing for tuberculosis should be considered in high-risk individuals. In those who are considered at low risk, tuberculosis testing may be done at the discretion of the dermatologist. In those with a history of HBV infection, HBV serologies should be pursued routinely given the risk for reactivation.
Annual screening for nonmelanoma skin cancer should be performed in all patients taking biologics. Tumor necrosis factor α inhibitor therapy in particular confers an elevated risk for cutaneous squamous cell carcinoma, especially in patients who are immunosuppressed at baseline and those with history of UV phototherapy. Use of acitretin alongside TNF-α inhibitors or ustekinumab may prevent squamous cell carcinoma formation in high-risk patients.
Because infliximab treatment poses an elevated risk of liver injury,11 liver function tests should be repeated 3 months following initiation of treatment and then every 6 to 12 months subsequently if results are normal. Periodic assessment of suicidal ideation is recommended in patients on brodalumab therapy, which may necessitate more frequent follow-up visits and potentially psychiatry referrals in certain patients. Patients taking IL-17 inhibitors, particularly those who are concurrently taking methotrexate, are at increased risk for developing mucocutaneous Candida infections; these patients should be monitored for such infections and treated appropriately.12
It is additionally important for prescribing dermatologists to ensure that patients on biologics are following up with their general providers to receive timely age-appropriate preventative screenings and vaccines. Inactivated vaccinations may be administered during therapy with any biologic; however, live vaccinations may induce systemic infection in those who are immunocompromised, which theoretically includes individuals taking biologic agents, though incidence data in this patient population are scarce.13 Some experts believe that administration of live vaccines warrants temporary discontinuation of biologic therapy for 2 to 3 half-lives before and after vaccination (Table). Others recommend stopping treatment at least 4 weeks before and until 2 weeks after vaccination. For patients taking biologics with half-lives greater than 20 days, which would theoretically require stopping the drug 2 months prior to vaccination, the benefit of vaccination should be weighed against the risk of prolonged discontinuation of therapy. Until recently, this recommendation was particularly important, as a live herpes zoster vaccination was recommended by the Centers for Disease Control and Prevention for adults older than 60 years. In 2017, a new inactivated herpes zoster vaccine was introduced and is now the preferred vaccine for all patients older than 50 years.14 It is especially important that patients on biologics receive this vaccine to avoid temporary drug discontinuation.
Evidence that any particular class of biologics increases risk for solid tumors or lymphoreticular malignancy is limited. One case-control analysis reported that more than 12 months of treatment with TNF-α inhibitors may increase risk for malignancy; however, the confidence interval reported hardly allows for statistical significance.15 Another retrospective cohort study found no elevated incidence of cancer in patients on TNF-α inhibitors compared to nonbiologic comparators.16 Ustekinumab was shown to confer no increased risk for malignancy in 1 large study,15 but no large studies have been conducted for other classes of drugs. Given the limited and inconclusive evidence available, the guidelines recommend that age-appropriate cancer screenings recommended for the general population should be pursued in patients taking biologics.
Surgery while taking biologics may lead to stress-induced augmentation of immunosuppression, resulting in elevated risk of infection.17 Low-risk surgeries that do not warrant discontinuation of treatment include endoscopic, ophthalmologic, dermatologic, orthopedic, and breast procedures. In patients preparing for elective surgery in which respiratory, gastrointestinal, or genitourinary tracts will be entered, biologics may be discontinued at least 3 half-lives (Table) prior to surgery if the dermatologist and surgeon collaboratively deem that risk of infection outweighs benefit of continued therapy.18 Therapy may be resumed within 1 to 2 weeks postoperatively if there are no surgical complications.
Switching Biologics
Changing therapy to another biologic should be considered if there is no response to treatment or the patient experiences adverse effects while taking a particular biologic. Because evidence is limited regarding the ideal time frame between discontinuation of a prior medication and initiation of a new biologic, this interval should be determined at the discretion of the provider based on the patient’s disease severity and response to prior treatment. For individuals who experience primary or secondary treatment failure while maintaining appropriate dosing and treatment compliance, switching to a different biologic is recommended to maximize treatment response.19 Changing therapy to a biologic within the same class is generally effective,20 and switching to a biologic with another mechanism of action should be considered if a class-specific adverse effect is the major reason for altering the regimen. Nonetheless, some patients may be unresponsive to biologic changes. Further research is necessary to determine which biologics may be most effective when previously used biologics have failed and particular factors that may predispose patients to biologic unresponsiveness.
Resuming Biologic Treatment Following Cessation
In cases where therapy is discontinued for any reason, it may be necessary to repeat initiation dosing when resuming treatment. In patients with severe or flaring disease or if more than 3 to 4 half-lives have passed since the most recent dose, it may be necessary to restart therapy with the loading dose (Table). Unfortunately, restarting therapy may preclude some patients from experiencing the maximal response that they attained prior to cessation. In such cases, switching biologic therapy to a different class may prove beneficial.
Final Thoughts
These recommendations contain valuable information that will assist dermatologists when initiating biologics and managing outcomes of their psoriasis patients. It is, however, crucial to bear in mind that these guidelines serve as merely a tool. Given the paucity of comprehensive research, particularly regarding some of the more recently approved therapies, there are many questions that are unanswered within the guidelines. Their utility for each individual patient situation is therefore limited, and clinical judgement may outweigh the information presented. The recommendations nevertheless provide a pivotal and unprecedented framework that promotes discourse among patients, dermatologists, and other providers to optimize the efficacy of biologic therapy for psoriasis.
- Michalek IM, Loring B, John SM. A systematic review of worldwide epidemiology of psoriasis. J Eur Acad Dermatol Venereol. 2017;31:205-212.
- Kurd SK, Gelfand JM. The prevalence of previously diagnosed and undiagnosed psoriasis in US adults: results from NHANES 2003-2004. J Am Acad Dermatol. 2009;60:218-224.
- Menter A, Strober BE, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics [published online February 13, 2019]. J Am Acad Dermatol. 2019;80:1029-1072.
- Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58:826-850.
- Förger F, Villiger PM. Treatment of rheumatoid arthritis during pregnancy: present and future. Expert Rev Clin Immunol. 2016;12:937-944.
- Gooderham M, Elewski B, Pariser D, et al. Incidence of serious gastrointestinal events and inflammatory bowel disease among tildrakizumab-treated patients with moderate-to-severe plaque psoriasis: data from 3 large randomized clinical trials [abstract]. J Am Acad Dermatol. 2018;79(suppl 1):AB166.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-328.
- Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br J Dermatol. 2016;175:273-286
- Beck KM, Koo J. Brodalumab for the treatment of plaque psoriasis: up-to-date. Expert Opin Biol Ther. 2019;19:287-292.
- Fouéré S, Adjadj L, Pawin H. How patients experience psoriasis: results from a European survey. J Eur Acad Dermatol Venereol. 2005;19(suppl 3):2-6.
- Björnsson ES, Bergmann OM, Björnsson HK, et al. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology. 2013;144:1419-1425, 1425.e1-3; quiz e19-20.
- Saunte DM, Mrowietz U, Puig L, et al. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br J Dermatol. 2017;177:47-62.
- Huber F, Ehrensperger B, Hatz C, et al. Safety of live vaccines on immunosuppressive or immunomodulatory therapy—a retrospective study in three Swiss Travel Clinics [published online January 1, 2018]. J Travel Med. doi:10.1093/jtm/tax082.
- Dooling KL, Guo A, Patel M, et al. Recommendations of the Advisory Committee on Immunization Practices for Use of Herpes Zoster Vaccines. MMWR Morb Mortal Wkly Rep. 2018;67:103-108.
- Fiorentino D, Ho V, Lebwohl MG, et al. Risk of malignancy with systemic psoriasis treatment in the Psoriasis Longitudinal Assessment Registry. J Am Acad Dermatol. 2017;77:845-854.e5.
- Haynes K, Beukelman T, Curtis JR, et al. Tumor necrosis factor α inhibitor therapy and cancer risk in chronic immune-mediated diseases. Arthritis Rheum. 2013;65:48-58.
- Fabiano A, De Simone C, Gisondi P, et al. Management of patients with psoriasis treated with biologic drugs needing a surgical treatment. Drug Dev Res. 2014;75(suppl 1):S24-S26.
- Choi YM, Debbaneh M, Weinberg JM, et al. From the Medical Board of the National Psoriasis Foundation: perioperative management of systemic immunomodulatory agents in patients with psoriasis and psoriatic arthritis. J Am Acad Dermatol. 2016;75:798-805.e7.
- Honda H, Umezawa Y, Kikuchi S, et al. Switching of biologics in psoriasis: reasons and results. J Dermatol. 2017;44:1015-1019.
- Bracke S, Lambert J. Viewpoint on handling anti-TNF failure in psoriasis. Arch Dermatol Res. 2013;305:945-950.
Psoriasis is a systemic immune-mediated disorder characterized by erythematous, scaly, well-demarcated plaques on the skin that affects approximately 3% of the world’s population.1 The disease is moderate to severe for approximately 1 in 6 individuals with psoriasis.2 These patients, particularly those with symptoms that are refractory to topical therapy and/or phototherapy, can benefit from the use of biologic agents, which are monoclonal antibodies and fusion proteins engineered to inhibit the action of cytokines that drive psoriatic inflammation.
In February 2019, the American Academy of Dermatology (AAD) and National Psoriasis Foundation (NPF) released an updated set of guidelines for the use of biologics in treating adult patients with psoriasis.3 The prior guidelines were released in 2008 when just 3 biologics—etanercept, infliximab, and adalimumab—were approved by the US Food and Drug Administration (FDA) for the management of psoriasis. These older recommendations were mostly based on studies of the efficacy and safety of biologics for patients with psoriatic arthritis.4 Over the last 11 years, 8 novel biologics have gained FDA approval, and numerous large phase 2 and phase 3 trials evaluating the risks and benefits of biologics have been conducted. The new guidelines contain considerably more detail and are based on evidence more specific to psoriasis rather than to psoriatic arthritis. Given the large repertoire of biologics available today and the increased amount of published research regarding each one, these guidelines may aid dermatologists in choosing the optimal biologic and managing therapy.
The AAD-NPF recommendations discuss the mechanism of action, efficacy, safety, and adverse events of the 10 biologics that have been FDA approved for the treatment of psoriasis as of March 2019, plus risankizumab, which was pending FDA approval at the time of publication and was later approved in April 2019. They also address dosing regimens, potential to combine biologics with other therapies, and different forms of psoriasis for which each may be effective.3 The purpose of this discussion is to present these guidelines in a condensed form to prescribers of biologic therapies and review the most clinically significant considerations during each step of treatment. Of note, we highlight only treatment of adult patients and do not discuss information relevant to risankizumab, as it was not FDA approved when the AAD-NPF guidelines were released.
Choosing a Biologic
Biologic therapy may be considered for patients with psoriasis that affects more than 3% of the body’s surface and is recalcitrant to localized therapies. There is no particular first-line biologic recommended for all patients with psoriasis; rather, choice of therapy should be individualized to the patient, considering factors such as body parts affected, comorbidities, lifestyle, and drug cost.
All 10 FDA-approved biologics (Table) have been ranked by the AAD and NPF as having grade A evidence for efficacy as monotherapy in the treatment of moderate to severe plaque-type psoriasis. Involvement of difficult-to-treat areas may be considered when choosing a specific therapy. The tumor necrosis factor α (TNF-α) inhibitors etanercept and adalimumab, the IL-17 inhibitor secukinumab, and the IL-23 inhibitor guselkumab have the greatest evidence for efficacy in treatment of nail disease. For scalp involvement, etanercept and guselkumab have the highest-quality evidence, and for palmoplantar disease, adalimumab, secukinumab, and guselkumab are considered the most effective. The TNF-α inhibitors are considered the optimal treatment option for concurrent psoriatic arthritis, though the IL-12/IL-23 inhibitor ustekinumab and the IL-17 inhibitors secukinumab and ixekizumab also have shown grade A evidence of efficacy. Of note, because TNF-α inhibitors received the earliest FDA approval, there is most evidence available for this class. Therapies with lower evidence quality for certain forms of psoriasis may show real-world effectiveness in individual patients, though more trials will be necessary to generate a body of evidence to change these clinical recommendations.
In pregnant women or those are anticipating pregnancy, certolizumab may be considered, as it is the only biologic shown to have minimal to no placental transfer. Other TNF-α inhibitors may undergo active placental transfer, particularly during the latter half of pregnancy,5 and the greatest theoretical risk of transfer occurs in the third trimester. Although these drugs may not directly harm the fetus, they do cause fetal immunosuppression for up to the first 3 months of life. All TNF-α inhibitors are considered safe during lactation. There are inadequate data regarding the safety of other classes of biologics during pregnancy and lactation.
Overweight and obese patients also require unique considerations when choosing a biologic. Infliximab is the only approved psoriasis biologic that utilizes proportional-to-weight dosing and hence may be particularly efficacious in patients with higher body mass. Ustekinumab dosing also takes patient weight into consideration; patients heavier than 100 kg should receive 90-mg doses at initiation and during maintenance compared to 45 mg for patients who weigh 100 kg or less. Other approved biologics also may be utilized in these patients but may require closer monitoring of treatment efficacy.
There are few serious contraindications for specific biologic therapies. Any history of allergic reaction to a particular therapy is an absolute contraindication to its use. In patients for whom IL-17 inhibitor treatment is being considered, inflammatory bowel disease (IBD) should be ruled out given the likelihood that IL-17 could reactivate or worsen IBD. Of note, TNF-α inhibitors and ustekinumab are approved therapies for patients with IBD and may be recommended in patients with comorbid psoriasis. Phase 2 and phase 3 trials have found no reactivation or worsening of IBD in patients with psoriasis who were treated with the IL-23 inhibitor tildrakizumab,6 and phase 2 trials of treatment of IBD with guselkumab are currently underway (ClinicalTrials.gov Identifier NCT03466411). In patients with New York Heart Association class III and class IV congestive heart failure or multiple sclerosis, initiation of TNF-α inhibitors should be avoided. Among 3 phase 3 trials encompassing nearly 3000 patients treated with the IL-17 inhibitor brodalumab, a total of 3 patients died by suicide7,8; hence, the FDA has issued a black box warning cautioning against use of this drug in patients with history of suicidal ideation or recent suicidal behavior. Although a causal relationship between brodalumab and suicide has not been well established,9 a thorough psychiatric history should be obtained in those initiating treatment with brodalumab.
Initiation of Therapy
Prior to initiating biologic therapy, it is important to obtain a complete blood cell count, complete metabolic panel, tuberculosis testing, and hepatitis B virus (HBV) and hepatitis C virus serologies. Testing for human immunodeficiency virus may be pursued at the clinician’s discretion. It is important to address any positive or concerning results prior to starting biologics. In patients with active infections, therapy may be initiated alongside guidance from an infectious disease specialist. Those with a positive purified protein derivative test, T-SPOT test, or QuantiFERON-TB Gold test must be referred for chest radiographs to rule out active tuberculosis. Patients with active HBV infection should receive appropriate referral to initiate antiviral therapy as well as core antibody testing, and those with active hepatitis C virus infection may only receive biologics under the combined discretion of a dermatologist and an appropriate specialist. Patients with human immunodeficiency virus must concurrently receive highly active antiretroviral therapy, show normal CD4+ T-cell count and undetectable viral load, and have no recent history of opportunistic infection.
Therapy should be commenced using specific dosing regimens, which are unique for each biologic (Table). Patients also must be educated on routine follow-up to assess treatment response and tolerability.
Assessment and Optimization of Treatment Response
Patients taking biologics may experience primary treatment failure, defined as lack of response to therapy from initiation. One predisposing factor may be increased body mass; patients who are overweight and obese are less likely to respond to standard regimens of TNF-α inhibitors and 45-mg dosing of ustekinumab. In most cases, however, the cause of primary nonresponse is unpredictable. For patients in whom therapy has failed within the recommended initial time frame (Table), dose escalation or shortening of dosing intervals may be pursued. Recommended dosing adjustments are outlined in the Table. Alternatively, patients may be switched to a different biologic.
If desired effectiveness is not reached with biologic monotherapy, topical corticosteroids, topical vitamin D analogues, or narrowband UVB light therapy may be concurrently used for difficult-to-treat areas. Evidence for safety and effectiveness of systemic adjuncts to biologics is moderate to low, warranting caution with their use. Methotrexate, cyclosporine, and apremilast have synergistic effects with biologics, though they may increase the risk for immunosuppression-related complications. Acitretin, an oral retinoid, likely is the most reasonable systemic adjunct to biologics because of its lack of immunosuppressive properties.
In patients with a suboptimal response to biologics, particularly those taking therapies that require frequent dosing, poor compliance should be considered.10 These patients may be switched to a biologic with less-frequent maintenance dosing (Table). Ustekinumab and tildrakizumab may be the best options for optimizing compliance, as they require dosing only once every 12 weeks after administration of loading doses.
Secondary treatment failure is diminished efficacy of treatment following successful initial response despite no changes in regimen. The best-known factor contributing to secondary nonresponse to biologics is the development of antidrug antibodies (ADAs), a phenomenon known as immunogenicity. The development of efficacy-limiting ADAs has been observed in response to most biologics, though ADAs against etanercept and guselkumab do not limit therapeutic response. Patients taking adalimumab and infliximab have particularly well-documented efficacy-limiting immunogenicity, and those who develop ADAs to infliximab are considered more prone to developing infusion reactions. Methotrexate, which limits antibody formation, may concomitantly be prescribed in patients who experience secondary treatment failure. It should be considered in all patients taking infliximab to increase efficacy and tolerability of therapy.
Considerations During Active Therapy
In addition to monitoring adherence and response to regimens, dermatologists must be heavily involved in counseling patients regarding the risks and adverse effects associated with these therapies. During maintenance therapy with biologics, patients must follow up with the prescriber at minimum every 3 to 6 months to evaluate for continued efficacy of treatment, extent of side effects, and effects of treatment on overall health and quality of life. Given the immunosuppressive effects of biologics, annual testing for tuberculosis should be considered in high-risk individuals. In those who are considered at low risk, tuberculosis testing may be done at the discretion of the dermatologist. In those with a history of HBV infection, HBV serologies should be pursued routinely given the risk for reactivation.
Annual screening for nonmelanoma skin cancer should be performed in all patients taking biologics. Tumor necrosis factor α inhibitor therapy in particular confers an elevated risk for cutaneous squamous cell carcinoma, especially in patients who are immunosuppressed at baseline and those with history of UV phototherapy. Use of acitretin alongside TNF-α inhibitors or ustekinumab may prevent squamous cell carcinoma formation in high-risk patients.
Because infliximab treatment poses an elevated risk of liver injury,11 liver function tests should be repeated 3 months following initiation of treatment and then every 6 to 12 months subsequently if results are normal. Periodic assessment of suicidal ideation is recommended in patients on brodalumab therapy, which may necessitate more frequent follow-up visits and potentially psychiatry referrals in certain patients. Patients taking IL-17 inhibitors, particularly those who are concurrently taking methotrexate, are at increased risk for developing mucocutaneous Candida infections; these patients should be monitored for such infections and treated appropriately.12
It is additionally important for prescribing dermatologists to ensure that patients on biologics are following up with their general providers to receive timely age-appropriate preventative screenings and vaccines. Inactivated vaccinations may be administered during therapy with any biologic; however, live vaccinations may induce systemic infection in those who are immunocompromised, which theoretically includes individuals taking biologic agents, though incidence data in this patient population are scarce.13 Some experts believe that administration of live vaccines warrants temporary discontinuation of biologic therapy for 2 to 3 half-lives before and after vaccination (Table). Others recommend stopping treatment at least 4 weeks before and until 2 weeks after vaccination. For patients taking biologics with half-lives greater than 20 days, which would theoretically require stopping the drug 2 months prior to vaccination, the benefit of vaccination should be weighed against the risk of prolonged discontinuation of therapy. Until recently, this recommendation was particularly important, as a live herpes zoster vaccination was recommended by the Centers for Disease Control and Prevention for adults older than 60 years. In 2017, a new inactivated herpes zoster vaccine was introduced and is now the preferred vaccine for all patients older than 50 years.14 It is especially important that patients on biologics receive this vaccine to avoid temporary drug discontinuation.
Evidence that any particular class of biologics increases risk for solid tumors or lymphoreticular malignancy is limited. One case-control analysis reported that more than 12 months of treatment with TNF-α inhibitors may increase risk for malignancy; however, the confidence interval reported hardly allows for statistical significance.15 Another retrospective cohort study found no elevated incidence of cancer in patients on TNF-α inhibitors compared to nonbiologic comparators.16 Ustekinumab was shown to confer no increased risk for malignancy in 1 large study,15 but no large studies have been conducted for other classes of drugs. Given the limited and inconclusive evidence available, the guidelines recommend that age-appropriate cancer screenings recommended for the general population should be pursued in patients taking biologics.
Surgery while taking biologics may lead to stress-induced augmentation of immunosuppression, resulting in elevated risk of infection.17 Low-risk surgeries that do not warrant discontinuation of treatment include endoscopic, ophthalmologic, dermatologic, orthopedic, and breast procedures. In patients preparing for elective surgery in which respiratory, gastrointestinal, or genitourinary tracts will be entered, biologics may be discontinued at least 3 half-lives (Table) prior to surgery if the dermatologist and surgeon collaboratively deem that risk of infection outweighs benefit of continued therapy.18 Therapy may be resumed within 1 to 2 weeks postoperatively if there are no surgical complications.
Switching Biologics
Changing therapy to another biologic should be considered if there is no response to treatment or the patient experiences adverse effects while taking a particular biologic. Because evidence is limited regarding the ideal time frame between discontinuation of a prior medication and initiation of a new biologic, this interval should be determined at the discretion of the provider based on the patient’s disease severity and response to prior treatment. For individuals who experience primary or secondary treatment failure while maintaining appropriate dosing and treatment compliance, switching to a different biologic is recommended to maximize treatment response.19 Changing therapy to a biologic within the same class is generally effective,20 and switching to a biologic with another mechanism of action should be considered if a class-specific adverse effect is the major reason for altering the regimen. Nonetheless, some patients may be unresponsive to biologic changes. Further research is necessary to determine which biologics may be most effective when previously used biologics have failed and particular factors that may predispose patients to biologic unresponsiveness.
Resuming Biologic Treatment Following Cessation
In cases where therapy is discontinued for any reason, it may be necessary to repeat initiation dosing when resuming treatment. In patients with severe or flaring disease or if more than 3 to 4 half-lives have passed since the most recent dose, it may be necessary to restart therapy with the loading dose (Table). Unfortunately, restarting therapy may preclude some patients from experiencing the maximal response that they attained prior to cessation. In such cases, switching biologic therapy to a different class may prove beneficial.
Final Thoughts
These recommendations contain valuable information that will assist dermatologists when initiating biologics and managing outcomes of their psoriasis patients. It is, however, crucial to bear in mind that these guidelines serve as merely a tool. Given the paucity of comprehensive research, particularly regarding some of the more recently approved therapies, there are many questions that are unanswered within the guidelines. Their utility for each individual patient situation is therefore limited, and clinical judgement may outweigh the information presented. The recommendations nevertheless provide a pivotal and unprecedented framework that promotes discourse among patients, dermatologists, and other providers to optimize the efficacy of biologic therapy for psoriasis.
Psoriasis is a systemic immune-mediated disorder characterized by erythematous, scaly, well-demarcated plaques on the skin that affects approximately 3% of the world’s population.1 The disease is moderate to severe for approximately 1 in 6 individuals with psoriasis.2 These patients, particularly those with symptoms that are refractory to topical therapy and/or phototherapy, can benefit from the use of biologic agents, which are monoclonal antibodies and fusion proteins engineered to inhibit the action of cytokines that drive psoriatic inflammation.
In February 2019, the American Academy of Dermatology (AAD) and National Psoriasis Foundation (NPF) released an updated set of guidelines for the use of biologics in treating adult patients with psoriasis.3 The prior guidelines were released in 2008 when just 3 biologics—etanercept, infliximab, and adalimumab—were approved by the US Food and Drug Administration (FDA) for the management of psoriasis. These older recommendations were mostly based on studies of the efficacy and safety of biologics for patients with psoriatic arthritis.4 Over the last 11 years, 8 novel biologics have gained FDA approval, and numerous large phase 2 and phase 3 trials evaluating the risks and benefits of biologics have been conducted. The new guidelines contain considerably more detail and are based on evidence more specific to psoriasis rather than to psoriatic arthritis. Given the large repertoire of biologics available today and the increased amount of published research regarding each one, these guidelines may aid dermatologists in choosing the optimal biologic and managing therapy.
The AAD-NPF recommendations discuss the mechanism of action, efficacy, safety, and adverse events of the 10 biologics that have been FDA approved for the treatment of psoriasis as of March 2019, plus risankizumab, which was pending FDA approval at the time of publication and was later approved in April 2019. They also address dosing regimens, potential to combine biologics with other therapies, and different forms of psoriasis for which each may be effective.3 The purpose of this discussion is to present these guidelines in a condensed form to prescribers of biologic therapies and review the most clinically significant considerations during each step of treatment. Of note, we highlight only treatment of adult patients and do not discuss information relevant to risankizumab, as it was not FDA approved when the AAD-NPF guidelines were released.
Choosing a Biologic
Biologic therapy may be considered for patients with psoriasis that affects more than 3% of the body’s surface and is recalcitrant to localized therapies. There is no particular first-line biologic recommended for all patients with psoriasis; rather, choice of therapy should be individualized to the patient, considering factors such as body parts affected, comorbidities, lifestyle, and drug cost.
All 10 FDA-approved biologics (Table) have been ranked by the AAD and NPF as having grade A evidence for efficacy as monotherapy in the treatment of moderate to severe plaque-type psoriasis. Involvement of difficult-to-treat areas may be considered when choosing a specific therapy. The tumor necrosis factor α (TNF-α) inhibitors etanercept and adalimumab, the IL-17 inhibitor secukinumab, and the IL-23 inhibitor guselkumab have the greatest evidence for efficacy in treatment of nail disease. For scalp involvement, etanercept and guselkumab have the highest-quality evidence, and for palmoplantar disease, adalimumab, secukinumab, and guselkumab are considered the most effective. The TNF-α inhibitors are considered the optimal treatment option for concurrent psoriatic arthritis, though the IL-12/IL-23 inhibitor ustekinumab and the IL-17 inhibitors secukinumab and ixekizumab also have shown grade A evidence of efficacy. Of note, because TNF-α inhibitors received the earliest FDA approval, there is most evidence available for this class. Therapies with lower evidence quality for certain forms of psoriasis may show real-world effectiveness in individual patients, though more trials will be necessary to generate a body of evidence to change these clinical recommendations.
In pregnant women or those are anticipating pregnancy, certolizumab may be considered, as it is the only biologic shown to have minimal to no placental transfer. Other TNF-α inhibitors may undergo active placental transfer, particularly during the latter half of pregnancy,5 and the greatest theoretical risk of transfer occurs in the third trimester. Although these drugs may not directly harm the fetus, they do cause fetal immunosuppression for up to the first 3 months of life. All TNF-α inhibitors are considered safe during lactation. There are inadequate data regarding the safety of other classes of biologics during pregnancy and lactation.
Overweight and obese patients also require unique considerations when choosing a biologic. Infliximab is the only approved psoriasis biologic that utilizes proportional-to-weight dosing and hence may be particularly efficacious in patients with higher body mass. Ustekinumab dosing also takes patient weight into consideration; patients heavier than 100 kg should receive 90-mg doses at initiation and during maintenance compared to 45 mg for patients who weigh 100 kg or less. Other approved biologics also may be utilized in these patients but may require closer monitoring of treatment efficacy.
There are few serious contraindications for specific biologic therapies. Any history of allergic reaction to a particular therapy is an absolute contraindication to its use. In patients for whom IL-17 inhibitor treatment is being considered, inflammatory bowel disease (IBD) should be ruled out given the likelihood that IL-17 could reactivate or worsen IBD. Of note, TNF-α inhibitors and ustekinumab are approved therapies for patients with IBD and may be recommended in patients with comorbid psoriasis. Phase 2 and phase 3 trials have found no reactivation or worsening of IBD in patients with psoriasis who were treated with the IL-23 inhibitor tildrakizumab,6 and phase 2 trials of treatment of IBD with guselkumab are currently underway (ClinicalTrials.gov Identifier NCT03466411). In patients with New York Heart Association class III and class IV congestive heart failure or multiple sclerosis, initiation of TNF-α inhibitors should be avoided. Among 3 phase 3 trials encompassing nearly 3000 patients treated with the IL-17 inhibitor brodalumab, a total of 3 patients died by suicide7,8; hence, the FDA has issued a black box warning cautioning against use of this drug in patients with history of suicidal ideation or recent suicidal behavior. Although a causal relationship between brodalumab and suicide has not been well established,9 a thorough psychiatric history should be obtained in those initiating treatment with brodalumab.
Initiation of Therapy
Prior to initiating biologic therapy, it is important to obtain a complete blood cell count, complete metabolic panel, tuberculosis testing, and hepatitis B virus (HBV) and hepatitis C virus serologies. Testing for human immunodeficiency virus may be pursued at the clinician’s discretion. It is important to address any positive or concerning results prior to starting biologics. In patients with active infections, therapy may be initiated alongside guidance from an infectious disease specialist. Those with a positive purified protein derivative test, T-SPOT test, or QuantiFERON-TB Gold test must be referred for chest radiographs to rule out active tuberculosis. Patients with active HBV infection should receive appropriate referral to initiate antiviral therapy as well as core antibody testing, and those with active hepatitis C virus infection may only receive biologics under the combined discretion of a dermatologist and an appropriate specialist. Patients with human immunodeficiency virus must concurrently receive highly active antiretroviral therapy, show normal CD4+ T-cell count and undetectable viral load, and have no recent history of opportunistic infection.
Therapy should be commenced using specific dosing regimens, which are unique for each biologic (Table). Patients also must be educated on routine follow-up to assess treatment response and tolerability.
Assessment and Optimization of Treatment Response
Patients taking biologics may experience primary treatment failure, defined as lack of response to therapy from initiation. One predisposing factor may be increased body mass; patients who are overweight and obese are less likely to respond to standard regimens of TNF-α inhibitors and 45-mg dosing of ustekinumab. In most cases, however, the cause of primary nonresponse is unpredictable. For patients in whom therapy has failed within the recommended initial time frame (Table), dose escalation or shortening of dosing intervals may be pursued. Recommended dosing adjustments are outlined in the Table. Alternatively, patients may be switched to a different biologic.
If desired effectiveness is not reached with biologic monotherapy, topical corticosteroids, topical vitamin D analogues, or narrowband UVB light therapy may be concurrently used for difficult-to-treat areas. Evidence for safety and effectiveness of systemic adjuncts to biologics is moderate to low, warranting caution with their use. Methotrexate, cyclosporine, and apremilast have synergistic effects with biologics, though they may increase the risk for immunosuppression-related complications. Acitretin, an oral retinoid, likely is the most reasonable systemic adjunct to biologics because of its lack of immunosuppressive properties.
In patients with a suboptimal response to biologics, particularly those taking therapies that require frequent dosing, poor compliance should be considered.10 These patients may be switched to a biologic with less-frequent maintenance dosing (Table). Ustekinumab and tildrakizumab may be the best options for optimizing compliance, as they require dosing only once every 12 weeks after administration of loading doses.
Secondary treatment failure is diminished efficacy of treatment following successful initial response despite no changes in regimen. The best-known factor contributing to secondary nonresponse to biologics is the development of antidrug antibodies (ADAs), a phenomenon known as immunogenicity. The development of efficacy-limiting ADAs has been observed in response to most biologics, though ADAs against etanercept and guselkumab do not limit therapeutic response. Patients taking adalimumab and infliximab have particularly well-documented efficacy-limiting immunogenicity, and those who develop ADAs to infliximab are considered more prone to developing infusion reactions. Methotrexate, which limits antibody formation, may concomitantly be prescribed in patients who experience secondary treatment failure. It should be considered in all patients taking infliximab to increase efficacy and tolerability of therapy.
Considerations During Active Therapy
In addition to monitoring adherence and response to regimens, dermatologists must be heavily involved in counseling patients regarding the risks and adverse effects associated with these therapies. During maintenance therapy with biologics, patients must follow up with the prescriber at minimum every 3 to 6 months to evaluate for continued efficacy of treatment, extent of side effects, and effects of treatment on overall health and quality of life. Given the immunosuppressive effects of biologics, annual testing for tuberculosis should be considered in high-risk individuals. In those who are considered at low risk, tuberculosis testing may be done at the discretion of the dermatologist. In those with a history of HBV infection, HBV serologies should be pursued routinely given the risk for reactivation.
Annual screening for nonmelanoma skin cancer should be performed in all patients taking biologics. Tumor necrosis factor α inhibitor therapy in particular confers an elevated risk for cutaneous squamous cell carcinoma, especially in patients who are immunosuppressed at baseline and those with history of UV phototherapy. Use of acitretin alongside TNF-α inhibitors or ustekinumab may prevent squamous cell carcinoma formation in high-risk patients.
Because infliximab treatment poses an elevated risk of liver injury,11 liver function tests should be repeated 3 months following initiation of treatment and then every 6 to 12 months subsequently if results are normal. Periodic assessment of suicidal ideation is recommended in patients on brodalumab therapy, which may necessitate more frequent follow-up visits and potentially psychiatry referrals in certain patients. Patients taking IL-17 inhibitors, particularly those who are concurrently taking methotrexate, are at increased risk for developing mucocutaneous Candida infections; these patients should be monitored for such infections and treated appropriately.12
It is additionally important for prescribing dermatologists to ensure that patients on biologics are following up with their general providers to receive timely age-appropriate preventative screenings and vaccines. Inactivated vaccinations may be administered during therapy with any biologic; however, live vaccinations may induce systemic infection in those who are immunocompromised, which theoretically includes individuals taking biologic agents, though incidence data in this patient population are scarce.13 Some experts believe that administration of live vaccines warrants temporary discontinuation of biologic therapy for 2 to 3 half-lives before and after vaccination (Table). Others recommend stopping treatment at least 4 weeks before and until 2 weeks after vaccination. For patients taking biologics with half-lives greater than 20 days, which would theoretically require stopping the drug 2 months prior to vaccination, the benefit of vaccination should be weighed against the risk of prolonged discontinuation of therapy. Until recently, this recommendation was particularly important, as a live herpes zoster vaccination was recommended by the Centers for Disease Control and Prevention for adults older than 60 years. In 2017, a new inactivated herpes zoster vaccine was introduced and is now the preferred vaccine for all patients older than 50 years.14 It is especially important that patients on biologics receive this vaccine to avoid temporary drug discontinuation.
Evidence that any particular class of biologics increases risk for solid tumors or lymphoreticular malignancy is limited. One case-control analysis reported that more than 12 months of treatment with TNF-α inhibitors may increase risk for malignancy; however, the confidence interval reported hardly allows for statistical significance.15 Another retrospective cohort study found no elevated incidence of cancer in patients on TNF-α inhibitors compared to nonbiologic comparators.16 Ustekinumab was shown to confer no increased risk for malignancy in 1 large study,15 but no large studies have been conducted for other classes of drugs. Given the limited and inconclusive evidence available, the guidelines recommend that age-appropriate cancer screenings recommended for the general population should be pursued in patients taking biologics.
Surgery while taking biologics may lead to stress-induced augmentation of immunosuppression, resulting in elevated risk of infection.17 Low-risk surgeries that do not warrant discontinuation of treatment include endoscopic, ophthalmologic, dermatologic, orthopedic, and breast procedures. In patients preparing for elective surgery in which respiratory, gastrointestinal, or genitourinary tracts will be entered, biologics may be discontinued at least 3 half-lives (Table) prior to surgery if the dermatologist and surgeon collaboratively deem that risk of infection outweighs benefit of continued therapy.18 Therapy may be resumed within 1 to 2 weeks postoperatively if there are no surgical complications.
Switching Biologics
Changing therapy to another biologic should be considered if there is no response to treatment or the patient experiences adverse effects while taking a particular biologic. Because evidence is limited regarding the ideal time frame between discontinuation of a prior medication and initiation of a new biologic, this interval should be determined at the discretion of the provider based on the patient’s disease severity and response to prior treatment. For individuals who experience primary or secondary treatment failure while maintaining appropriate dosing and treatment compliance, switching to a different biologic is recommended to maximize treatment response.19 Changing therapy to a biologic within the same class is generally effective,20 and switching to a biologic with another mechanism of action should be considered if a class-specific adverse effect is the major reason for altering the regimen. Nonetheless, some patients may be unresponsive to biologic changes. Further research is necessary to determine which biologics may be most effective when previously used biologics have failed and particular factors that may predispose patients to biologic unresponsiveness.
Resuming Biologic Treatment Following Cessation
In cases where therapy is discontinued for any reason, it may be necessary to repeat initiation dosing when resuming treatment. In patients with severe or flaring disease or if more than 3 to 4 half-lives have passed since the most recent dose, it may be necessary to restart therapy with the loading dose (Table). Unfortunately, restarting therapy may preclude some patients from experiencing the maximal response that they attained prior to cessation. In such cases, switching biologic therapy to a different class may prove beneficial.
Final Thoughts
These recommendations contain valuable information that will assist dermatologists when initiating biologics and managing outcomes of their psoriasis patients. It is, however, crucial to bear in mind that these guidelines serve as merely a tool. Given the paucity of comprehensive research, particularly regarding some of the more recently approved therapies, there are many questions that are unanswered within the guidelines. Their utility for each individual patient situation is therefore limited, and clinical judgement may outweigh the information presented. The recommendations nevertheless provide a pivotal and unprecedented framework that promotes discourse among patients, dermatologists, and other providers to optimize the efficacy of biologic therapy for psoriasis.
- Michalek IM, Loring B, John SM. A systematic review of worldwide epidemiology of psoriasis. J Eur Acad Dermatol Venereol. 2017;31:205-212.
- Kurd SK, Gelfand JM. The prevalence of previously diagnosed and undiagnosed psoriasis in US adults: results from NHANES 2003-2004. J Am Acad Dermatol. 2009;60:218-224.
- Menter A, Strober BE, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics [published online February 13, 2019]. J Am Acad Dermatol. 2019;80:1029-1072.
- Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58:826-850.
- Förger F, Villiger PM. Treatment of rheumatoid arthritis during pregnancy: present and future. Expert Rev Clin Immunol. 2016;12:937-944.
- Gooderham M, Elewski B, Pariser D, et al. Incidence of serious gastrointestinal events and inflammatory bowel disease among tildrakizumab-treated patients with moderate-to-severe plaque psoriasis: data from 3 large randomized clinical trials [abstract]. J Am Acad Dermatol. 2018;79(suppl 1):AB166.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-328.
- Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br J Dermatol. 2016;175:273-286
- Beck KM, Koo J. Brodalumab for the treatment of plaque psoriasis: up-to-date. Expert Opin Biol Ther. 2019;19:287-292.
- Fouéré S, Adjadj L, Pawin H. How patients experience psoriasis: results from a European survey. J Eur Acad Dermatol Venereol. 2005;19(suppl 3):2-6.
- Björnsson ES, Bergmann OM, Björnsson HK, et al. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology. 2013;144:1419-1425, 1425.e1-3; quiz e19-20.
- Saunte DM, Mrowietz U, Puig L, et al. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br J Dermatol. 2017;177:47-62.
- Huber F, Ehrensperger B, Hatz C, et al. Safety of live vaccines on immunosuppressive or immunomodulatory therapy—a retrospective study in three Swiss Travel Clinics [published online January 1, 2018]. J Travel Med. doi:10.1093/jtm/tax082.
- Dooling KL, Guo A, Patel M, et al. Recommendations of the Advisory Committee on Immunization Practices for Use of Herpes Zoster Vaccines. MMWR Morb Mortal Wkly Rep. 2018;67:103-108.
- Fiorentino D, Ho V, Lebwohl MG, et al. Risk of malignancy with systemic psoriasis treatment in the Psoriasis Longitudinal Assessment Registry. J Am Acad Dermatol. 2017;77:845-854.e5.
- Haynes K, Beukelman T, Curtis JR, et al. Tumor necrosis factor α inhibitor therapy and cancer risk in chronic immune-mediated diseases. Arthritis Rheum. 2013;65:48-58.
- Fabiano A, De Simone C, Gisondi P, et al. Management of patients with psoriasis treated with biologic drugs needing a surgical treatment. Drug Dev Res. 2014;75(suppl 1):S24-S26.
- Choi YM, Debbaneh M, Weinberg JM, et al. From the Medical Board of the National Psoriasis Foundation: perioperative management of systemic immunomodulatory agents in patients with psoriasis and psoriatic arthritis. J Am Acad Dermatol. 2016;75:798-805.e7.
- Honda H, Umezawa Y, Kikuchi S, et al. Switching of biologics in psoriasis: reasons and results. J Dermatol. 2017;44:1015-1019.
- Bracke S, Lambert J. Viewpoint on handling anti-TNF failure in psoriasis. Arch Dermatol Res. 2013;305:945-950.
- Michalek IM, Loring B, John SM. A systematic review of worldwide epidemiology of psoriasis. J Eur Acad Dermatol Venereol. 2017;31:205-212.
- Kurd SK, Gelfand JM. The prevalence of previously diagnosed and undiagnosed psoriasis in US adults: results from NHANES 2003-2004. J Am Acad Dermatol. 2009;60:218-224.
- Menter A, Strober BE, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics [published online February 13, 2019]. J Am Acad Dermatol. 2019;80:1029-1072.
- Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58:826-850.
- Förger F, Villiger PM. Treatment of rheumatoid arthritis during pregnancy: present and future. Expert Rev Clin Immunol. 2016;12:937-944.
- Gooderham M, Elewski B, Pariser D, et al. Incidence of serious gastrointestinal events and inflammatory bowel disease among tildrakizumab-treated patients with moderate-to-severe plaque psoriasis: data from 3 large randomized clinical trials [abstract]. J Am Acad Dermatol. 2018;79(suppl 1):AB166.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-328.
- Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br J Dermatol. 2016;175:273-286
- Beck KM, Koo J. Brodalumab for the treatment of plaque psoriasis: up-to-date. Expert Opin Biol Ther. 2019;19:287-292.
- Fouéré S, Adjadj L, Pawin H. How patients experience psoriasis: results from a European survey. J Eur Acad Dermatol Venereol. 2005;19(suppl 3):2-6.
- Björnsson ES, Bergmann OM, Björnsson HK, et al. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology. 2013;144:1419-1425, 1425.e1-3; quiz e19-20.
- Saunte DM, Mrowietz U, Puig L, et al. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br J Dermatol. 2017;177:47-62.
- Huber F, Ehrensperger B, Hatz C, et al. Safety of live vaccines on immunosuppressive or immunomodulatory therapy—a retrospective study in three Swiss Travel Clinics [published online January 1, 2018]. J Travel Med. doi:10.1093/jtm/tax082.
- Dooling KL, Guo A, Patel M, et al. Recommendations of the Advisory Committee on Immunization Practices for Use of Herpes Zoster Vaccines. MMWR Morb Mortal Wkly Rep. 2018;67:103-108.
- Fiorentino D, Ho V, Lebwohl MG, et al. Risk of malignancy with systemic psoriasis treatment in the Psoriasis Longitudinal Assessment Registry. J Am Acad Dermatol. 2017;77:845-854.e5.
- Haynes K, Beukelman T, Curtis JR, et al. Tumor necrosis factor α inhibitor therapy and cancer risk in chronic immune-mediated diseases. Arthritis Rheum. 2013;65:48-58.
- Fabiano A, De Simone C, Gisondi P, et al. Management of patients with psoriasis treated with biologic drugs needing a surgical treatment. Drug Dev Res. 2014;75(suppl 1):S24-S26.
- Choi YM, Debbaneh M, Weinberg JM, et al. From the Medical Board of the National Psoriasis Foundation: perioperative management of systemic immunomodulatory agents in patients with psoriasis and psoriatic arthritis. J Am Acad Dermatol. 2016;75:798-805.e7.
- Honda H, Umezawa Y, Kikuchi S, et al. Switching of biologics in psoriasis: reasons and results. J Dermatol. 2017;44:1015-1019.
- Bracke S, Lambert J. Viewpoint on handling anti-TNF failure in psoriasis. Arch Dermatol Res. 2013;305:945-950.
Practice Points
- There are currently 11 biologics approved for psoriasis, but there is no first-line or optimalbiologic. The choice must be made using clinical judgment based on a variety of medical and social factors.
- Frequent assessment for efficacy of and adverse events due to biologic therapy is warranted, as lack of response, loss of response, or severe side effects may warrant addition of concurrent therapies or switching to a different biologic.
- There are important considerations to make when immunizing and planning for surgery in patients on biologics.
Systemic Therapies in Psoriasis: An Update on Newly Approved and Pipeline Biologics and Oral Treatments
Recent advances in our understanding of psoriatic immune pathways have led to new generations of targeted therapies developed over the last 5 years. Although the pathogenesis of psoriasis remains to be fully elucidated, the success of these targeted therapies has confirmed a critical role of the IL-23/helper T cell (TH17) axis in maintaining the psoriatic immune cascade, a positive feedback loop in which IL-17, IL-12, and IL-23 released from myeloid dendritic cells lead to activation of helperT cells. Activated helper T cells—namely TH1, TH17, and TH22—release IL-17, IL-22, and other proinflammatory cytokines, amplifying the immune response and leading to keratinocyte proliferation and immune cell migration to psoriatic lesions. Inhibition of IL-17 and IL-23 by several biologics disrupts this aberrant inflammatory cascade and has led to dramatic improvements in outcomes, particularly among patients with moderate to severe disease.
Numerous biologics targeting these pathways and several oral treatments have been approved by the US Food and Drug Administration (FDA) for the treatment of psoriasis; in addition, a number of promising therapies are on the horizon, and knowledge of these medications might help guide our treatment approach to the patient with psoriasis. This article provides an update on the most recent (as of 2019) approved therapies and medications in the pipeline for moderate to severe plaque psoriasis, with a focus on systemic agents in phase 3 clinical trials. (Medications targeting psoriatic arthritis, biosimilars, and existing medications approved by the FDA prior to 2019 will not be discussed.)
Risankizumab
Risankizumab-rzaa (formerly BI 655066) is a humanized IgG1 monoclonal antibody that targets the p19 subunit of IL-23, selectively inhibiting the role of this critical cytokine in psoriatic inflammation.
Phase 1 Trial
In a phase 1 proof-of-concept study, 39 patients with moderate to severe plaque psoriasis received varying dosages of intravenous or subcutaneous risankizumab or placebo.1 At week 12, the percentage of risankizumab-treated patients achieving reduction in the psoriasis area and severity index (PASI) score by 75% (PASI 75), 90% (PASI 90), and 100% (PASI 100) was 87% (27/31; P<.001 vs placebo), 58% (18/31; P=.007 vs placebo), and 16% (5/31; P=.590 vs placebo), respectively. Improvements in PASI scores were observed as early as week 2. Adverse events (AEs) were reported by 65% of the risankizumab group and 88% of the placebo group. Serious AEs were reported in 4 patients receiving risankizumab, none of which were considered related to the study medication.1
Phase 2 Trial
A phase 2 comparator trial demonstrated noninferiority at higher dosages of risankizumab in comparison to the IL-12/IL-23 inhibitor ustekinumab.2 Among 166 participants with moderate to severe plaque psoriasis, PASI 90 at week 12 was met by 77% of participants receiving 90 or 180 mg of risankizumab compared to 40% receiving ustekinumab (P<.001). Onset of activity with risankizumab was faster and the duration of effect longer vs ustekinumab; by week 8, at least PASI 75 was achieved by approximately 80% of participants in the 90-mg and 180-mg risankizumab groups compared to 60% in the ustekinumab group; PASI score reductions generally were maintained for as long as 20 weeks after the final dose of risankizumab was administered.2
Phase 3 Trials
The 52-week UltIMMa-1 and UltIMMa-2 phase 3 trials compared subcutaneous risankizumab (150 mg) to ustekinumab (45 or 90 mg [weight-based dosing]) or placebo administered at weeks 0, 4, 16, 28, and 40 in approximately 1000 patients with moderate to severe plaque psoriasis.3 Patients initially assigned to placebo switched to risankizumab 150 mg at week 16. At week 16, PASI 90 was achieved by 75.3% of risankizumab-treated patients, 42.0% of ustekinumab-treated patients, and 4.9% of placebo-treated patients in UltIMMa-1, and by 74.8% of risankizumab-treated patients, 47.5% of ustekinumab-treated patients, and 2.0% of placebo-treated patients in UltIMMa-2 (P<.0001 vs placebo and ustekinumab for both studies). Achievement of a static physician’s global assessment (sPGA) score of 0 or 1 at week 16 similarly favored risankizumab, with 87.8%, 63.0%, and 7.8% of patients in UltIMMa-1 meeting an sPGA score of 0 or 1 in the risankizumab, ustekinumab, and placebo groups, respectively, and 83.7%, 61.6%, and 5.1% in UltIMMa-2 meeting an sPGA score of 0 or 1 in the risankizumab, ustekinumab, and placebo groups, respectively (P<.0001 vs placebo and ustekinumab for both studies). Among patients initially assigned to risankizumab, improvements in PASI and sPGA continued to increase until week 52, with 81.9% achieving PASI 90 at week 52 compared to 44.0% on ustekinumab in UltIMMa-1, and 80.6% achieving PASI 90 at week 52 compared to 50.5% on ustekinumab in UltIMMa-2 (P<.0001 vs ustekinumab for both studies). Treatment-emergent AE profiles were similar for risankizumab and ustekinumab in both studies, and there were no unexpected safety findings.3
Risankizumab received FDA approval for the treatment of moderate to severe plaque psoriasis in April 2019.
Bimekizumab
Bimekizumab (UCB4940), a humanized IgG1 monoclonal antibody, selectively neutralizes the biologic functions of IL-17A and IL-17F, the latter of which has only recently been implicated in contributing to the psoriatic immune cascade.4
First-in-Human Study
Thirty-nine participants with mild psoriasis demonstrated efficacy after single-dose intravenous bimekizumab, with maximal improvements in all measures of disease activity observed between weeks 8 and 12 in participants receiving 160 to 640 mg.5
Proof-of-Concept Phase 1b Study
A subsequent trial of 53 participants with psoriatic arthritis demonstrated sustained efficacy to week 20 with varying dosages of intravenous bimekizumab.6 At week 8, PASI 100 was met by 86.7% of participants receiving the top 3 dosages of bimekizumab compared to none of the placebo-treated participants. Treatment-emergent AEs, including neutropenia and elevation of liver transaminases, were mostly mild to moderate and resolved spontaneously. There were 3 severe AEs and 3 serious AEs, none of which were related to treatment.6
Importantly, bimekizumab was shown in this small study to have the potential to be highly effective at treating psoriatic arthritis. American College of Rheumatology ACR20, ACR50, and ACR70 response criteria were very high, with an ACR20 of 80% and an ACR50 of 40%.6 Further trials are necessary to gather more data and confirm these findings; however, these levels of response are higher than those of any other biologic on the market.
Phase 2b Dose-Ranging Study
In this trial, 250 participants with moderate to severe plaque psoriasis received either 64 mg, 160 mg with a 320-mg loading dose, 320 mg, or 480 mg of subcutaneous bimekizumab or placebo at weeks 0, 4, and 8.7 At week 12, PASI 90 was achieved by significantly more patients in all bimekizumab-treated groups compared to the placebo group (46.2%–79.1% vs 0%; P<.0001 for all dosages); PASI 100 also was achieved by significantly more bimekizumab-treated patients (27.9%–60.0% vs 0%; P<.0002). Improvement began as early as week 4, with clinically meaningful responses observed in all bimekizumab groups across all measures of disease activity. Treatment-emergent AEs occurred more frequently in bimekizumab-treated participants (61%) than in placebo-treated participants (36%); the most common AEs were nasopharyngitis and upper respiratory tract infection. Of note, fungal infections were reported by 4.3% of participants receiving bimekizumab; all cases were localized superficial infection, and none led to discontinuation. Three serious AEs were reported, none of which were considered related to the study treatment.7
Mirikizumab
Mirikizumab (LY3074828) is a humanized IgG4 monoclonal antibody that selectively binds and inhibits the p19 subunit of IL-23, with no action on IL-12.
Phase 1 Trial
Mirikizumab was shown to improve PASI scores in patients with plaque psoriasis.8
Phase 2 Trial
Subsequently, a trial of 205 participants with moderate to severe plaque psoriasis compared 3 dosing regimens of subcutaneous mirikizumab—30, 100, or 300 mg—at weeks 0 and 8 compared to placebo.9 Primary end point results at week 16 demonstrated PASI 90 response rates of 0%, 29% (P=.009), 59% (P<.001), and 67% (P<.001) in the placebo, 30-mg, 100-mg, and 300-mg mirikizumab groups, respectively. Complete clearance of psoriasis, measured by PASI 100 and sPGA 0, was achieved by 0%, 16%, 31%, and 31%, respectively (P=.039 for 30 mg vs placebo; P=.007 for the higher dosage groups vs placebo). Response rates for all efficacy outcomes were statistically significantly higher for all mirikizumab treatment groups compared to placebo and were highest in the 100-mg and 300-mg treatment groups. Frequencies of participants reporting AEs were similar across treatment and placebo groups.9
Oral Medications
Only a few small-molecule, orally bioavailable therapies are on the market for the treatment of psoriasis, some of which are associated with unfavorable side-effect profiles that preclude long-term therapy.
BMS-986165
The intracellular signaling enzyme tyrosine kinase 2 is involved in functional responses of IL-12 and IL-23. BMS-986165, a potent oral inhibitor of tyrosine kinase 2 with greater selectivity than other tyrosine kinase inhibitors, demonstrated efficacy in a phase 2 trial of 267 participants with moderate to severe plaque psoriasis receiving any of 5 dosing regimens—3 mg every other day, 3 mg daily, 3 mg twice daily, 6 mg twice daily, and 12 mg daily—compared to placebo.10 At week 12, the percentage of patients with a 75% or greater reduction in PASI was 7% with placebo, 9% with 3 mg every other day (P=.49 vs placebo), 39% with 3 mg daily (P<.001 vs placebo), 69% with 3 mg twice daily (P<.001 vs placebo), 67% with 6 mg twice daily (P<.001 vs placebo), and 75% with 12 mg once daily (P<.001 vs placebo). Adverse events occurred in 51% of patients in the placebo group and in 55% to 80% of BMS-986165–treated patients; the most common AEs were nasopharyngitis, headache, diarrhea, nausea, and upper respiratory tract infection.10
A phase 3 trial comparing BMS-986165 with placebo and apremilast is underway (ClinicalTrials.gov Identifier NCT03611751).
Piclidenoson (CF101)
A novel small molecule that binds the Gi protein–associated A3 adenosine receptor piclidenoson induces an anti-inflammatory response via deregulation of the Wnt and nuclear factor κB signal transduction pathways, leading to downregulation of proinflammatory cytokines, including IL-17 and IL-23.11
In a phase 2 dose-ranging study, 75 patients with moderate to severe plaque psoriasis received varying dosages—1, 2, or 4 mg—of oral piclidenoson or placebo twice daily for 12 weeks.12 Progressive improvement in the mean change from baseline PASI score was observed in the 2-mg group, with statistically significant differences at weeks 8 and 12 compared to placebo (P=.047 and P=.031, respectively). At week 12, 35.3% of the 2-mg group achieved at least PASI 50. Improvements in PASI were less pronounced in the 4-mg group, and no therapeutic benefit was observed in the 1-mg group. Of the 20 AEs reported, 15 possibly were related to the study drug; 1 AE was severe.12
In a subsequent phase 2/3 trial, patients with moderate to severe plaque psoriasis received piclidenoson—1 or 2 mg—or placebo twice daily.13 At week 12, PASI 75 was achieved by 8.5% of patients in the 2-mg group and by 6.9% of patients receiving placebo (P=.621), thereby not meeting the primary study end point. Results at week 32 were more encouraging. In the 2-mg group, PASI mean percentage improvement was 57% (P<.002) compared to baseline, with linear improvements observed in PASI 50 (63.5%), PASI 75 (35.5%), PASI 90 (24.7%), and PASI 100 (10.6%).13
A phase 3 trial comparing piclidenoson 2 and 3 mg to apremilast and placebo is in progress (ClinicalTrials.gov Identifier NCT03168256).
Future Directions
Despite abundant options for treating moderate to severe plaque psoriasis and psoriatic arthritis, the pipeline remains rich. Novel treatments might have improved efficacy, favorable safety profiles, and different modes of administration compared to current medications. In addition to the novel therapeutics covered here, several treatments are in development further down the pipeline, with only phase 1 or 2 data available. Remtolumab (ABT-122), a tumor necrosis factor α– and IL-17A–targeted immunoglobulin, is unique among biologics, given its dual inhibition of tumor necrosis factor α and IL-17A.14 M1095 (ALX-0761), a novel trivalent bispecific nanobody, is another intriguing candidate. This dual inhibitor of IL-17A/F might exhibit a number of advantages over conventional antibodies, including better tissue penetration, reduced immunogenicity, and a longer half-life (ClinicalTrials.gov Identifier NCT03384745).15,16
As always with drug development, numerous medications that were under development failed to meet primary end points in phase 2 trials and have therefore been discontinued, including namilumab and prurisol. It is reassuring that the pace of drug discovery and development in psoriasis does not seem to be slowing; to our patients’ benefit, we will have an array of treatments available to tailor therapy to the individual.
- Krueger JG, Ferris LK, Menter A, et al. Anti-IL-23A mAb BI 655066 for treatment of moderate-to-severe psoriasis: safety, efficacy, pharmacokinetics, and biomarker results of a single-rising-dose, randomized, double-blind, placebo-controlled trial. J Allergy Clin Immunol. 2015;136:116-124.e7.
- Papp KA, Blauvelt A, Bukhalo M, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. 2017;376:1551-1560.
- Gordon KB, Strober B, Lebwohl M, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392:650-661.
- Maroof A, Baeten D, Archer S, et al. 02.13 Il-17f contributes to human chronic inflammation in synovial tissue: preclinical evidence with dual IL-17a and IL-17f inhibition with bimekizumab in psoriatic arthritis. Ann Rheum Dis. 2017;76(Suppl 1):A13.
- Glatt S, Helmer E, Haier B, et al. First-in-human randomized study of bimekizumab, a humanized monoclonal antibody and selective dual inhibitor of IL-17A and IL-17F, in mild psoriasis. Br J Clin Pharmacol. 2017;83:991-1001.
- Glatt S, Baeten D, Baker T, et al. Dual IL-17A and IL-17F neutralisation by bimekizumab in psoriatic arthritis: evidence from preclinical experiments and a randomised placebo-controlled clinical trial that IL-17F contributes to human chronic tissue inflammation. Ann Rheum Dis. 2018;77:523-532.
- Papp KA, Merola JF, Gottlieb AB, et al. Dual neutralization of bothinterleukin 17A and interleukin 17F with bimekizumab in patients with psoriasis: results from BE ABLE 1, a 12-week randomized, double-blinded, placebo-controlled phase 2b trial. J Am Acad Dermatol. 2018;79:277-286.e10.
- Maari C. Safety, efficacy, and pharmacokinetics of a p19-directed IL-23 antibody in patients with plaque psoriasis and healthy subjects. Presented at: 25th European Academy of Dermatology and Venereology Congress; Vienna, Austria; September 28-October 2, 2016.
- Reich K, Rich P, Maari C, et al. Efficacy and safety of mirikizumab (LY3074828) in the treatment of moderate-to-severe plaque psoriasis: results from a randomized phase II study. Br J Dermatol. 2019;181:88-95.
- Papp K, Gordon K, Thaçi D, et al. Phase 2 trial of selective tyrosine kinase 2 inhibition in psoriasis. N Engl J Med. 2018;379:1313-1321.
- Cohen S, Barer F, Itzhak I, et al. Inhibition of IL-17 and IL-23 in human keratinocytes by the A3 adenosine receptor agonist piclidenoson. J Immunol Res. 2018;2018:2310970.
- David M, Akerman L, Ziv M, et al. Treatment of plaque-type psoriasis with oral CF101: data from an exploratory randomized phase 2 clinical trial. J Eur Acad Dermatol Venereol. 2012;26:361-367.
- 13. David M, Gospodinov DK, Gheorghe N, et al. Treatment of plaque-type psoriasis with oral CF101: data from a phase II/III multicenter, randomized, controlled trial. J Drugs Dermatol. 2016;15:931-938.
- Mease PJ, Genovese MC, Weinblatt ME, et al. Phase II study of ABT-122, a tumor necrosis factor- and interleukin-17A-targeted dual variable domain immunoglobulin, in patients with psoriatic arthritis with an inadequate response to methotrexate. Arthritis Rheumatol. 2018;70:1778-1789.
- Nanobodies’ competitive features. Ablynx website. http://www.ablynx.com/technology-innovation/nanobodies-competitive-features. Accessed July 4, 2019.
- Svecova D, Lubell MW, Casset-Semanaz F, et al. A randomized, double-blind, placebo-controlled phase 1 study of multiple ascending doses of subcutaneous M1095, an anti-interleukin-17A/F nanobody, in moderate-to-severe psoriasis. J Am Acad Dermatol. 2019;81:196-203.
Recent advances in our understanding of psoriatic immune pathways have led to new generations of targeted therapies developed over the last 5 years. Although the pathogenesis of psoriasis remains to be fully elucidated, the success of these targeted therapies has confirmed a critical role of the IL-23/helper T cell (TH17) axis in maintaining the psoriatic immune cascade, a positive feedback loop in which IL-17, IL-12, and IL-23 released from myeloid dendritic cells lead to activation of helperT cells. Activated helper T cells—namely TH1, TH17, and TH22—release IL-17, IL-22, and other proinflammatory cytokines, amplifying the immune response and leading to keratinocyte proliferation and immune cell migration to psoriatic lesions. Inhibition of IL-17 and IL-23 by several biologics disrupts this aberrant inflammatory cascade and has led to dramatic improvements in outcomes, particularly among patients with moderate to severe disease.
Numerous biologics targeting these pathways and several oral treatments have been approved by the US Food and Drug Administration (FDA) for the treatment of psoriasis; in addition, a number of promising therapies are on the horizon, and knowledge of these medications might help guide our treatment approach to the patient with psoriasis. This article provides an update on the most recent (as of 2019) approved therapies and medications in the pipeline for moderate to severe plaque psoriasis, with a focus on systemic agents in phase 3 clinical trials. (Medications targeting psoriatic arthritis, biosimilars, and existing medications approved by the FDA prior to 2019 will not be discussed.)
Risankizumab
Risankizumab-rzaa (formerly BI 655066) is a humanized IgG1 monoclonal antibody that targets the p19 subunit of IL-23, selectively inhibiting the role of this critical cytokine in psoriatic inflammation.
Phase 1 Trial
In a phase 1 proof-of-concept study, 39 patients with moderate to severe plaque psoriasis received varying dosages of intravenous or subcutaneous risankizumab or placebo.1 At week 12, the percentage of risankizumab-treated patients achieving reduction in the psoriasis area and severity index (PASI) score by 75% (PASI 75), 90% (PASI 90), and 100% (PASI 100) was 87% (27/31; P<.001 vs placebo), 58% (18/31; P=.007 vs placebo), and 16% (5/31; P=.590 vs placebo), respectively. Improvements in PASI scores were observed as early as week 2. Adverse events (AEs) were reported by 65% of the risankizumab group and 88% of the placebo group. Serious AEs were reported in 4 patients receiving risankizumab, none of which were considered related to the study medication.1
Phase 2 Trial
A phase 2 comparator trial demonstrated noninferiority at higher dosages of risankizumab in comparison to the IL-12/IL-23 inhibitor ustekinumab.2 Among 166 participants with moderate to severe plaque psoriasis, PASI 90 at week 12 was met by 77% of participants receiving 90 or 180 mg of risankizumab compared to 40% receiving ustekinumab (P<.001). Onset of activity with risankizumab was faster and the duration of effect longer vs ustekinumab; by week 8, at least PASI 75 was achieved by approximately 80% of participants in the 90-mg and 180-mg risankizumab groups compared to 60% in the ustekinumab group; PASI score reductions generally were maintained for as long as 20 weeks after the final dose of risankizumab was administered.2
Phase 3 Trials
The 52-week UltIMMa-1 and UltIMMa-2 phase 3 trials compared subcutaneous risankizumab (150 mg) to ustekinumab (45 or 90 mg [weight-based dosing]) or placebo administered at weeks 0, 4, 16, 28, and 40 in approximately 1000 patients with moderate to severe plaque psoriasis.3 Patients initially assigned to placebo switched to risankizumab 150 mg at week 16. At week 16, PASI 90 was achieved by 75.3% of risankizumab-treated patients, 42.0% of ustekinumab-treated patients, and 4.9% of placebo-treated patients in UltIMMa-1, and by 74.8% of risankizumab-treated patients, 47.5% of ustekinumab-treated patients, and 2.0% of placebo-treated patients in UltIMMa-2 (P<.0001 vs placebo and ustekinumab for both studies). Achievement of a static physician’s global assessment (sPGA) score of 0 or 1 at week 16 similarly favored risankizumab, with 87.8%, 63.0%, and 7.8% of patients in UltIMMa-1 meeting an sPGA score of 0 or 1 in the risankizumab, ustekinumab, and placebo groups, respectively, and 83.7%, 61.6%, and 5.1% in UltIMMa-2 meeting an sPGA score of 0 or 1 in the risankizumab, ustekinumab, and placebo groups, respectively (P<.0001 vs placebo and ustekinumab for both studies). Among patients initially assigned to risankizumab, improvements in PASI and sPGA continued to increase until week 52, with 81.9% achieving PASI 90 at week 52 compared to 44.0% on ustekinumab in UltIMMa-1, and 80.6% achieving PASI 90 at week 52 compared to 50.5% on ustekinumab in UltIMMa-2 (P<.0001 vs ustekinumab for both studies). Treatment-emergent AE profiles were similar for risankizumab and ustekinumab in both studies, and there were no unexpected safety findings.3
Risankizumab received FDA approval for the treatment of moderate to severe plaque psoriasis in April 2019.
Bimekizumab
Bimekizumab (UCB4940), a humanized IgG1 monoclonal antibody, selectively neutralizes the biologic functions of IL-17A and IL-17F, the latter of which has only recently been implicated in contributing to the psoriatic immune cascade.4
First-in-Human Study
Thirty-nine participants with mild psoriasis demonstrated efficacy after single-dose intravenous bimekizumab, with maximal improvements in all measures of disease activity observed between weeks 8 and 12 in participants receiving 160 to 640 mg.5
Proof-of-Concept Phase 1b Study
A subsequent trial of 53 participants with psoriatic arthritis demonstrated sustained efficacy to week 20 with varying dosages of intravenous bimekizumab.6 At week 8, PASI 100 was met by 86.7% of participants receiving the top 3 dosages of bimekizumab compared to none of the placebo-treated participants. Treatment-emergent AEs, including neutropenia and elevation of liver transaminases, were mostly mild to moderate and resolved spontaneously. There were 3 severe AEs and 3 serious AEs, none of which were related to treatment.6
Importantly, bimekizumab was shown in this small study to have the potential to be highly effective at treating psoriatic arthritis. American College of Rheumatology ACR20, ACR50, and ACR70 response criteria were very high, with an ACR20 of 80% and an ACR50 of 40%.6 Further trials are necessary to gather more data and confirm these findings; however, these levels of response are higher than those of any other biologic on the market.
Phase 2b Dose-Ranging Study
In this trial, 250 participants with moderate to severe plaque psoriasis received either 64 mg, 160 mg with a 320-mg loading dose, 320 mg, or 480 mg of subcutaneous bimekizumab or placebo at weeks 0, 4, and 8.7 At week 12, PASI 90 was achieved by significantly more patients in all bimekizumab-treated groups compared to the placebo group (46.2%–79.1% vs 0%; P<.0001 for all dosages); PASI 100 also was achieved by significantly more bimekizumab-treated patients (27.9%–60.0% vs 0%; P<.0002). Improvement began as early as week 4, with clinically meaningful responses observed in all bimekizumab groups across all measures of disease activity. Treatment-emergent AEs occurred more frequently in bimekizumab-treated participants (61%) than in placebo-treated participants (36%); the most common AEs were nasopharyngitis and upper respiratory tract infection. Of note, fungal infections were reported by 4.3% of participants receiving bimekizumab; all cases were localized superficial infection, and none led to discontinuation. Three serious AEs were reported, none of which were considered related to the study treatment.7
Mirikizumab
Mirikizumab (LY3074828) is a humanized IgG4 monoclonal antibody that selectively binds and inhibits the p19 subunit of IL-23, with no action on IL-12.
Phase 1 Trial
Mirikizumab was shown to improve PASI scores in patients with plaque psoriasis.8
Phase 2 Trial
Subsequently, a trial of 205 participants with moderate to severe plaque psoriasis compared 3 dosing regimens of subcutaneous mirikizumab—30, 100, or 300 mg—at weeks 0 and 8 compared to placebo.9 Primary end point results at week 16 demonstrated PASI 90 response rates of 0%, 29% (P=.009), 59% (P<.001), and 67% (P<.001) in the placebo, 30-mg, 100-mg, and 300-mg mirikizumab groups, respectively. Complete clearance of psoriasis, measured by PASI 100 and sPGA 0, was achieved by 0%, 16%, 31%, and 31%, respectively (P=.039 for 30 mg vs placebo; P=.007 for the higher dosage groups vs placebo). Response rates for all efficacy outcomes were statistically significantly higher for all mirikizumab treatment groups compared to placebo and were highest in the 100-mg and 300-mg treatment groups. Frequencies of participants reporting AEs were similar across treatment and placebo groups.9
Oral Medications
Only a few small-molecule, orally bioavailable therapies are on the market for the treatment of psoriasis, some of which are associated with unfavorable side-effect profiles that preclude long-term therapy.
BMS-986165
The intracellular signaling enzyme tyrosine kinase 2 is involved in functional responses of IL-12 and IL-23. BMS-986165, a potent oral inhibitor of tyrosine kinase 2 with greater selectivity than other tyrosine kinase inhibitors, demonstrated efficacy in a phase 2 trial of 267 participants with moderate to severe plaque psoriasis receiving any of 5 dosing regimens—3 mg every other day, 3 mg daily, 3 mg twice daily, 6 mg twice daily, and 12 mg daily—compared to placebo.10 At week 12, the percentage of patients with a 75% or greater reduction in PASI was 7% with placebo, 9% with 3 mg every other day (P=.49 vs placebo), 39% with 3 mg daily (P<.001 vs placebo), 69% with 3 mg twice daily (P<.001 vs placebo), 67% with 6 mg twice daily (P<.001 vs placebo), and 75% with 12 mg once daily (P<.001 vs placebo). Adverse events occurred in 51% of patients in the placebo group and in 55% to 80% of BMS-986165–treated patients; the most common AEs were nasopharyngitis, headache, diarrhea, nausea, and upper respiratory tract infection.10
A phase 3 trial comparing BMS-986165 with placebo and apremilast is underway (ClinicalTrials.gov Identifier NCT03611751).
Piclidenoson (CF101)
A novel small molecule that binds the Gi protein–associated A3 adenosine receptor piclidenoson induces an anti-inflammatory response via deregulation of the Wnt and nuclear factor κB signal transduction pathways, leading to downregulation of proinflammatory cytokines, including IL-17 and IL-23.11
In a phase 2 dose-ranging study, 75 patients with moderate to severe plaque psoriasis received varying dosages—1, 2, or 4 mg—of oral piclidenoson or placebo twice daily for 12 weeks.12 Progressive improvement in the mean change from baseline PASI score was observed in the 2-mg group, with statistically significant differences at weeks 8 and 12 compared to placebo (P=.047 and P=.031, respectively). At week 12, 35.3% of the 2-mg group achieved at least PASI 50. Improvements in PASI were less pronounced in the 4-mg group, and no therapeutic benefit was observed in the 1-mg group. Of the 20 AEs reported, 15 possibly were related to the study drug; 1 AE was severe.12
In a subsequent phase 2/3 trial, patients with moderate to severe plaque psoriasis received piclidenoson—1 or 2 mg—or placebo twice daily.13 At week 12, PASI 75 was achieved by 8.5% of patients in the 2-mg group and by 6.9% of patients receiving placebo (P=.621), thereby not meeting the primary study end point. Results at week 32 were more encouraging. In the 2-mg group, PASI mean percentage improvement was 57% (P<.002) compared to baseline, with linear improvements observed in PASI 50 (63.5%), PASI 75 (35.5%), PASI 90 (24.7%), and PASI 100 (10.6%).13
A phase 3 trial comparing piclidenoson 2 and 3 mg to apremilast and placebo is in progress (ClinicalTrials.gov Identifier NCT03168256).
Future Directions
Despite abundant options for treating moderate to severe plaque psoriasis and psoriatic arthritis, the pipeline remains rich. Novel treatments might have improved efficacy, favorable safety profiles, and different modes of administration compared to current medications. In addition to the novel therapeutics covered here, several treatments are in development further down the pipeline, with only phase 1 or 2 data available. Remtolumab (ABT-122), a tumor necrosis factor α– and IL-17A–targeted immunoglobulin, is unique among biologics, given its dual inhibition of tumor necrosis factor α and IL-17A.14 M1095 (ALX-0761), a novel trivalent bispecific nanobody, is another intriguing candidate. This dual inhibitor of IL-17A/F might exhibit a number of advantages over conventional antibodies, including better tissue penetration, reduced immunogenicity, and a longer half-life (ClinicalTrials.gov Identifier NCT03384745).15,16
As always with drug development, numerous medications that were under development failed to meet primary end points in phase 2 trials and have therefore been discontinued, including namilumab and prurisol. It is reassuring that the pace of drug discovery and development in psoriasis does not seem to be slowing; to our patients’ benefit, we will have an array of treatments available to tailor therapy to the individual.
Recent advances in our understanding of psoriatic immune pathways have led to new generations of targeted therapies developed over the last 5 years. Although the pathogenesis of psoriasis remains to be fully elucidated, the success of these targeted therapies has confirmed a critical role of the IL-23/helper T cell (TH17) axis in maintaining the psoriatic immune cascade, a positive feedback loop in which IL-17, IL-12, and IL-23 released from myeloid dendritic cells lead to activation of helperT cells. Activated helper T cells—namely TH1, TH17, and TH22—release IL-17, IL-22, and other proinflammatory cytokines, amplifying the immune response and leading to keratinocyte proliferation and immune cell migration to psoriatic lesions. Inhibition of IL-17 and IL-23 by several biologics disrupts this aberrant inflammatory cascade and has led to dramatic improvements in outcomes, particularly among patients with moderate to severe disease.
Numerous biologics targeting these pathways and several oral treatments have been approved by the US Food and Drug Administration (FDA) for the treatment of psoriasis; in addition, a number of promising therapies are on the horizon, and knowledge of these medications might help guide our treatment approach to the patient with psoriasis. This article provides an update on the most recent (as of 2019) approved therapies and medications in the pipeline for moderate to severe plaque psoriasis, with a focus on systemic agents in phase 3 clinical trials. (Medications targeting psoriatic arthritis, biosimilars, and existing medications approved by the FDA prior to 2019 will not be discussed.)
Risankizumab
Risankizumab-rzaa (formerly BI 655066) is a humanized IgG1 monoclonal antibody that targets the p19 subunit of IL-23, selectively inhibiting the role of this critical cytokine in psoriatic inflammation.
Phase 1 Trial
In a phase 1 proof-of-concept study, 39 patients with moderate to severe plaque psoriasis received varying dosages of intravenous or subcutaneous risankizumab or placebo.1 At week 12, the percentage of risankizumab-treated patients achieving reduction in the psoriasis area and severity index (PASI) score by 75% (PASI 75), 90% (PASI 90), and 100% (PASI 100) was 87% (27/31; P<.001 vs placebo), 58% (18/31; P=.007 vs placebo), and 16% (5/31; P=.590 vs placebo), respectively. Improvements in PASI scores were observed as early as week 2. Adverse events (AEs) were reported by 65% of the risankizumab group and 88% of the placebo group. Serious AEs were reported in 4 patients receiving risankizumab, none of which were considered related to the study medication.1
Phase 2 Trial
A phase 2 comparator trial demonstrated noninferiority at higher dosages of risankizumab in comparison to the IL-12/IL-23 inhibitor ustekinumab.2 Among 166 participants with moderate to severe plaque psoriasis, PASI 90 at week 12 was met by 77% of participants receiving 90 or 180 mg of risankizumab compared to 40% receiving ustekinumab (P<.001). Onset of activity with risankizumab was faster and the duration of effect longer vs ustekinumab; by week 8, at least PASI 75 was achieved by approximately 80% of participants in the 90-mg and 180-mg risankizumab groups compared to 60% in the ustekinumab group; PASI score reductions generally were maintained for as long as 20 weeks after the final dose of risankizumab was administered.2
Phase 3 Trials
The 52-week UltIMMa-1 and UltIMMa-2 phase 3 trials compared subcutaneous risankizumab (150 mg) to ustekinumab (45 or 90 mg [weight-based dosing]) or placebo administered at weeks 0, 4, 16, 28, and 40 in approximately 1000 patients with moderate to severe plaque psoriasis.3 Patients initially assigned to placebo switched to risankizumab 150 mg at week 16. At week 16, PASI 90 was achieved by 75.3% of risankizumab-treated patients, 42.0% of ustekinumab-treated patients, and 4.9% of placebo-treated patients in UltIMMa-1, and by 74.8% of risankizumab-treated patients, 47.5% of ustekinumab-treated patients, and 2.0% of placebo-treated patients in UltIMMa-2 (P<.0001 vs placebo and ustekinumab for both studies). Achievement of a static physician’s global assessment (sPGA) score of 0 or 1 at week 16 similarly favored risankizumab, with 87.8%, 63.0%, and 7.8% of patients in UltIMMa-1 meeting an sPGA score of 0 or 1 in the risankizumab, ustekinumab, and placebo groups, respectively, and 83.7%, 61.6%, and 5.1% in UltIMMa-2 meeting an sPGA score of 0 or 1 in the risankizumab, ustekinumab, and placebo groups, respectively (P<.0001 vs placebo and ustekinumab for both studies). Among patients initially assigned to risankizumab, improvements in PASI and sPGA continued to increase until week 52, with 81.9% achieving PASI 90 at week 52 compared to 44.0% on ustekinumab in UltIMMa-1, and 80.6% achieving PASI 90 at week 52 compared to 50.5% on ustekinumab in UltIMMa-2 (P<.0001 vs ustekinumab for both studies). Treatment-emergent AE profiles were similar for risankizumab and ustekinumab in both studies, and there were no unexpected safety findings.3
Risankizumab received FDA approval for the treatment of moderate to severe plaque psoriasis in April 2019.
Bimekizumab
Bimekizumab (UCB4940), a humanized IgG1 monoclonal antibody, selectively neutralizes the biologic functions of IL-17A and IL-17F, the latter of which has only recently been implicated in contributing to the psoriatic immune cascade.4
First-in-Human Study
Thirty-nine participants with mild psoriasis demonstrated efficacy after single-dose intravenous bimekizumab, with maximal improvements in all measures of disease activity observed between weeks 8 and 12 in participants receiving 160 to 640 mg.5
Proof-of-Concept Phase 1b Study
A subsequent trial of 53 participants with psoriatic arthritis demonstrated sustained efficacy to week 20 with varying dosages of intravenous bimekizumab.6 At week 8, PASI 100 was met by 86.7% of participants receiving the top 3 dosages of bimekizumab compared to none of the placebo-treated participants. Treatment-emergent AEs, including neutropenia and elevation of liver transaminases, were mostly mild to moderate and resolved spontaneously. There were 3 severe AEs and 3 serious AEs, none of which were related to treatment.6
Importantly, bimekizumab was shown in this small study to have the potential to be highly effective at treating psoriatic arthritis. American College of Rheumatology ACR20, ACR50, and ACR70 response criteria were very high, with an ACR20 of 80% and an ACR50 of 40%.6 Further trials are necessary to gather more data and confirm these findings; however, these levels of response are higher than those of any other biologic on the market.
Phase 2b Dose-Ranging Study
In this trial, 250 participants with moderate to severe plaque psoriasis received either 64 mg, 160 mg with a 320-mg loading dose, 320 mg, or 480 mg of subcutaneous bimekizumab or placebo at weeks 0, 4, and 8.7 At week 12, PASI 90 was achieved by significantly more patients in all bimekizumab-treated groups compared to the placebo group (46.2%–79.1% vs 0%; P<.0001 for all dosages); PASI 100 also was achieved by significantly more bimekizumab-treated patients (27.9%–60.0% vs 0%; P<.0002). Improvement began as early as week 4, with clinically meaningful responses observed in all bimekizumab groups across all measures of disease activity. Treatment-emergent AEs occurred more frequently in bimekizumab-treated participants (61%) than in placebo-treated participants (36%); the most common AEs were nasopharyngitis and upper respiratory tract infection. Of note, fungal infections were reported by 4.3% of participants receiving bimekizumab; all cases were localized superficial infection, and none led to discontinuation. Three serious AEs were reported, none of which were considered related to the study treatment.7
Mirikizumab
Mirikizumab (LY3074828) is a humanized IgG4 monoclonal antibody that selectively binds and inhibits the p19 subunit of IL-23, with no action on IL-12.
Phase 1 Trial
Mirikizumab was shown to improve PASI scores in patients with plaque psoriasis.8
Phase 2 Trial
Subsequently, a trial of 205 participants with moderate to severe plaque psoriasis compared 3 dosing regimens of subcutaneous mirikizumab—30, 100, or 300 mg—at weeks 0 and 8 compared to placebo.9 Primary end point results at week 16 demonstrated PASI 90 response rates of 0%, 29% (P=.009), 59% (P<.001), and 67% (P<.001) in the placebo, 30-mg, 100-mg, and 300-mg mirikizumab groups, respectively. Complete clearance of psoriasis, measured by PASI 100 and sPGA 0, was achieved by 0%, 16%, 31%, and 31%, respectively (P=.039 for 30 mg vs placebo; P=.007 for the higher dosage groups vs placebo). Response rates for all efficacy outcomes were statistically significantly higher for all mirikizumab treatment groups compared to placebo and were highest in the 100-mg and 300-mg treatment groups. Frequencies of participants reporting AEs were similar across treatment and placebo groups.9
Oral Medications
Only a few small-molecule, orally bioavailable therapies are on the market for the treatment of psoriasis, some of which are associated with unfavorable side-effect profiles that preclude long-term therapy.
BMS-986165
The intracellular signaling enzyme tyrosine kinase 2 is involved in functional responses of IL-12 and IL-23. BMS-986165, a potent oral inhibitor of tyrosine kinase 2 with greater selectivity than other tyrosine kinase inhibitors, demonstrated efficacy in a phase 2 trial of 267 participants with moderate to severe plaque psoriasis receiving any of 5 dosing regimens—3 mg every other day, 3 mg daily, 3 mg twice daily, 6 mg twice daily, and 12 mg daily—compared to placebo.10 At week 12, the percentage of patients with a 75% or greater reduction in PASI was 7% with placebo, 9% with 3 mg every other day (P=.49 vs placebo), 39% with 3 mg daily (P<.001 vs placebo), 69% with 3 mg twice daily (P<.001 vs placebo), 67% with 6 mg twice daily (P<.001 vs placebo), and 75% with 12 mg once daily (P<.001 vs placebo). Adverse events occurred in 51% of patients in the placebo group and in 55% to 80% of BMS-986165–treated patients; the most common AEs were nasopharyngitis, headache, diarrhea, nausea, and upper respiratory tract infection.10
A phase 3 trial comparing BMS-986165 with placebo and apremilast is underway (ClinicalTrials.gov Identifier NCT03611751).
Piclidenoson (CF101)
A novel small molecule that binds the Gi protein–associated A3 adenosine receptor piclidenoson induces an anti-inflammatory response via deregulation of the Wnt and nuclear factor κB signal transduction pathways, leading to downregulation of proinflammatory cytokines, including IL-17 and IL-23.11
In a phase 2 dose-ranging study, 75 patients with moderate to severe plaque psoriasis received varying dosages—1, 2, or 4 mg—of oral piclidenoson or placebo twice daily for 12 weeks.12 Progressive improvement in the mean change from baseline PASI score was observed in the 2-mg group, with statistically significant differences at weeks 8 and 12 compared to placebo (P=.047 and P=.031, respectively). At week 12, 35.3% of the 2-mg group achieved at least PASI 50. Improvements in PASI were less pronounced in the 4-mg group, and no therapeutic benefit was observed in the 1-mg group. Of the 20 AEs reported, 15 possibly were related to the study drug; 1 AE was severe.12
In a subsequent phase 2/3 trial, patients with moderate to severe plaque psoriasis received piclidenoson—1 or 2 mg—or placebo twice daily.13 At week 12, PASI 75 was achieved by 8.5% of patients in the 2-mg group and by 6.9% of patients receiving placebo (P=.621), thereby not meeting the primary study end point. Results at week 32 were more encouraging. In the 2-mg group, PASI mean percentage improvement was 57% (P<.002) compared to baseline, with linear improvements observed in PASI 50 (63.5%), PASI 75 (35.5%), PASI 90 (24.7%), and PASI 100 (10.6%).13
A phase 3 trial comparing piclidenoson 2 and 3 mg to apremilast and placebo is in progress (ClinicalTrials.gov Identifier NCT03168256).
Future Directions
Despite abundant options for treating moderate to severe plaque psoriasis and psoriatic arthritis, the pipeline remains rich. Novel treatments might have improved efficacy, favorable safety profiles, and different modes of administration compared to current medications. In addition to the novel therapeutics covered here, several treatments are in development further down the pipeline, with only phase 1 or 2 data available. Remtolumab (ABT-122), a tumor necrosis factor α– and IL-17A–targeted immunoglobulin, is unique among biologics, given its dual inhibition of tumor necrosis factor α and IL-17A.14 M1095 (ALX-0761), a novel trivalent bispecific nanobody, is another intriguing candidate. This dual inhibitor of IL-17A/F might exhibit a number of advantages over conventional antibodies, including better tissue penetration, reduced immunogenicity, and a longer half-life (ClinicalTrials.gov Identifier NCT03384745).15,16
As always with drug development, numerous medications that were under development failed to meet primary end points in phase 2 trials and have therefore been discontinued, including namilumab and prurisol. It is reassuring that the pace of drug discovery and development in psoriasis does not seem to be slowing; to our patients’ benefit, we will have an array of treatments available to tailor therapy to the individual.
- Krueger JG, Ferris LK, Menter A, et al. Anti-IL-23A mAb BI 655066 for treatment of moderate-to-severe psoriasis: safety, efficacy, pharmacokinetics, and biomarker results of a single-rising-dose, randomized, double-blind, placebo-controlled trial. J Allergy Clin Immunol. 2015;136:116-124.e7.
- Papp KA, Blauvelt A, Bukhalo M, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. 2017;376:1551-1560.
- Gordon KB, Strober B, Lebwohl M, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392:650-661.
- Maroof A, Baeten D, Archer S, et al. 02.13 Il-17f contributes to human chronic inflammation in synovial tissue: preclinical evidence with dual IL-17a and IL-17f inhibition with bimekizumab in psoriatic arthritis. Ann Rheum Dis. 2017;76(Suppl 1):A13.
- Glatt S, Helmer E, Haier B, et al. First-in-human randomized study of bimekizumab, a humanized monoclonal antibody and selective dual inhibitor of IL-17A and IL-17F, in mild psoriasis. Br J Clin Pharmacol. 2017;83:991-1001.
- Glatt S, Baeten D, Baker T, et al. Dual IL-17A and IL-17F neutralisation by bimekizumab in psoriatic arthritis: evidence from preclinical experiments and a randomised placebo-controlled clinical trial that IL-17F contributes to human chronic tissue inflammation. Ann Rheum Dis. 2018;77:523-532.
- Papp KA, Merola JF, Gottlieb AB, et al. Dual neutralization of bothinterleukin 17A and interleukin 17F with bimekizumab in patients with psoriasis: results from BE ABLE 1, a 12-week randomized, double-blinded, placebo-controlled phase 2b trial. J Am Acad Dermatol. 2018;79:277-286.e10.
- Maari C. Safety, efficacy, and pharmacokinetics of a p19-directed IL-23 antibody in patients with plaque psoriasis and healthy subjects. Presented at: 25th European Academy of Dermatology and Venereology Congress; Vienna, Austria; September 28-October 2, 2016.
- Reich K, Rich P, Maari C, et al. Efficacy and safety of mirikizumab (LY3074828) in the treatment of moderate-to-severe plaque psoriasis: results from a randomized phase II study. Br J Dermatol. 2019;181:88-95.
- Papp K, Gordon K, Thaçi D, et al. Phase 2 trial of selective tyrosine kinase 2 inhibition in psoriasis. N Engl J Med. 2018;379:1313-1321.
- Cohen S, Barer F, Itzhak I, et al. Inhibition of IL-17 and IL-23 in human keratinocytes by the A3 adenosine receptor agonist piclidenoson. J Immunol Res. 2018;2018:2310970.
- David M, Akerman L, Ziv M, et al. Treatment of plaque-type psoriasis with oral CF101: data from an exploratory randomized phase 2 clinical trial. J Eur Acad Dermatol Venereol. 2012;26:361-367.
- 13. David M, Gospodinov DK, Gheorghe N, et al. Treatment of plaque-type psoriasis with oral CF101: data from a phase II/III multicenter, randomized, controlled trial. J Drugs Dermatol. 2016;15:931-938.
- Mease PJ, Genovese MC, Weinblatt ME, et al. Phase II study of ABT-122, a tumor necrosis factor- and interleukin-17A-targeted dual variable domain immunoglobulin, in patients with psoriatic arthritis with an inadequate response to methotrexate. Arthritis Rheumatol. 2018;70:1778-1789.
- Nanobodies’ competitive features. Ablynx website. http://www.ablynx.com/technology-innovation/nanobodies-competitive-features. Accessed July 4, 2019.
- Svecova D, Lubell MW, Casset-Semanaz F, et al. A randomized, double-blind, placebo-controlled phase 1 study of multiple ascending doses of subcutaneous M1095, an anti-interleukin-17A/F nanobody, in moderate-to-severe psoriasis. J Am Acad Dermatol. 2019;81:196-203.
- Krueger JG, Ferris LK, Menter A, et al. Anti-IL-23A mAb BI 655066 for treatment of moderate-to-severe psoriasis: safety, efficacy, pharmacokinetics, and biomarker results of a single-rising-dose, randomized, double-blind, placebo-controlled trial. J Allergy Clin Immunol. 2015;136:116-124.e7.
- Papp KA, Blauvelt A, Bukhalo M, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. 2017;376:1551-1560.
- Gordon KB, Strober B, Lebwohl M, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392:650-661.
- Maroof A, Baeten D, Archer S, et al. 02.13 Il-17f contributes to human chronic inflammation in synovial tissue: preclinical evidence with dual IL-17a and IL-17f inhibition with bimekizumab in psoriatic arthritis. Ann Rheum Dis. 2017;76(Suppl 1):A13.
- Glatt S, Helmer E, Haier B, et al. First-in-human randomized study of bimekizumab, a humanized monoclonal antibody and selective dual inhibitor of IL-17A and IL-17F, in mild psoriasis. Br J Clin Pharmacol. 2017;83:991-1001.
- Glatt S, Baeten D, Baker T, et al. Dual IL-17A and IL-17F neutralisation by bimekizumab in psoriatic arthritis: evidence from preclinical experiments and a randomised placebo-controlled clinical trial that IL-17F contributes to human chronic tissue inflammation. Ann Rheum Dis. 2018;77:523-532.
- Papp KA, Merola JF, Gottlieb AB, et al. Dual neutralization of bothinterleukin 17A and interleukin 17F with bimekizumab in patients with psoriasis: results from BE ABLE 1, a 12-week randomized, double-blinded, placebo-controlled phase 2b trial. J Am Acad Dermatol. 2018;79:277-286.e10.
- Maari C. Safety, efficacy, and pharmacokinetics of a p19-directed IL-23 antibody in patients with plaque psoriasis and healthy subjects. Presented at: 25th European Academy of Dermatology and Venereology Congress; Vienna, Austria; September 28-October 2, 2016.
- Reich K, Rich P, Maari C, et al. Efficacy and safety of mirikizumab (LY3074828) in the treatment of moderate-to-severe plaque psoriasis: results from a randomized phase II study. Br J Dermatol. 2019;181:88-95.
- Papp K, Gordon K, Thaçi D, et al. Phase 2 trial of selective tyrosine kinase 2 inhibition in psoriasis. N Engl J Med. 2018;379:1313-1321.
- Cohen S, Barer F, Itzhak I, et al. Inhibition of IL-17 and IL-23 in human keratinocytes by the A3 adenosine receptor agonist piclidenoson. J Immunol Res. 2018;2018:2310970.
- David M, Akerman L, Ziv M, et al. Treatment of plaque-type psoriasis with oral CF101: data from an exploratory randomized phase 2 clinical trial. J Eur Acad Dermatol Venereol. 2012;26:361-367.
- 13. David M, Gospodinov DK, Gheorghe N, et al. Treatment of plaque-type psoriasis with oral CF101: data from a phase II/III multicenter, randomized, controlled trial. J Drugs Dermatol. 2016;15:931-938.
- Mease PJ, Genovese MC, Weinblatt ME, et al. Phase II study of ABT-122, a tumor necrosis factor- and interleukin-17A-targeted dual variable domain immunoglobulin, in patients with psoriatic arthritis with an inadequate response to methotrexate. Arthritis Rheumatol. 2018;70:1778-1789.
- Nanobodies’ competitive features. Ablynx website. http://www.ablynx.com/technology-innovation/nanobodies-competitive-features. Accessed July 4, 2019.
- Svecova D, Lubell MW, Casset-Semanaz F, et al. A randomized, double-blind, placebo-controlled phase 1 study of multiple ascending doses of subcutaneous M1095, an anti-interleukin-17A/F nanobody, in moderate-to-severe psoriasis. J Am Acad Dermatol. 2019;81:196-203.
Practice Points
- New systemic options for the treatment of psoriasis continue to emerge.
- With more choices, we can now tailor therapeutic approaches to the patient rather than base treatment choices purely on efficacy.
- New and upcoming biologics may offer improved skin clearance in line with the National Psoriasis Foundation’s treat-to-target approach, while others may offer increased efficacy in treating psoriatic arthritis.
- Novel small-molecule oral medications are in development and may have improved efficacy over current options.
ECT breaks super-refractory status epilepticus
BANGKOK –

“These were highly refractory patients and ECT did contribute to their status termination,” she reported at the congress, sponsored by the International League Against Epilepsy.
Dr. Nguyen, a neurologist at Oregon Health and Science University, Portland, presented a single-center retrospective case series composed of four ECT-treated patients with NORSE (new onset refractory status epilepticus), three of whom experienced marked improvement in their seizure activity, including cessation of their status epilepticus, after completing a course of eight or nine ECT sessions.
A four-patient series may not seem to be compelling evidence, but it’s a significant contribution to the sparse literature regarding this off-label usage of ECT. Indeed, the biggest case series to date consists of eight patients treated at Indiana University, five of whom displayed neurotelemetry or clinical evidence of improvement within 24 hours after completing a full course of ECT (J ECT. 2018 Mar;34[1]:e5-e9. doi: 10.1097/YCT.0000000000000450).
And realistically, these small case series are as good as the supporting evidence is likely to get.
“It would be incredibly difficult to perform a randomized trial, and even a case-control study in such refractory patients would be quite difficult,” she observed.
Super-refractory status epilepticus is the term for seizures persisting despite 24 hours of adequate treatment with benzodiazepines, loading doses of antiepileptic drugs, and anesthetic agents for medically-induced coma, or for seizures that resume after withdrawal of general anesthesia. NORSE is defined as refractory status epilepticus without a history of seizures or a readily identifiable etiology. It is often associated with an autoimmune or paraneoplastic encephalitis. Outcomes are generally poor.
The four ECT-treated patients in the Oregon series were aged 27-48 years. Two had a prodromal viral illness, and a third had a vague prodrome but a negative infectious disease workup. Three patients were on four antiepileptic drugs at the time they began ECT, one was on seven. Two patients had generalized seizures, while the other two had both generalized and focal seizures. Two had normal brain MRI scans and the other two had abnormal MRIs. The patients had CSF white blood cell counts of 2, 5, 10, and 58 per mm3. All patients were on immunotherapy with intravenous corticosteroids, and three of the four were on additional immunomodulatory drugs.
ECT was started on a compassionate-use basis 16-49 days after hospital admission. These were patients who had run out of options, according to Dr. Nguyen.
Three of the four patients returned to consciousness after completing their course of ECT and withdrawal of their general anesthesia, with attenuation of their seizure activity and cessation of their super-refractory status epilepticus. Two of the three were discharged to a rehabilitation facility, including one who eventually could ambulate with slight assistance. Another patient died after discharge, while the fourth patient died during the initial hospital stay due to septic shock unrelated to the ECT.
At discharge, one patient had a modified Rankin Scale score (mRS) of 4, and two had a score of 5. At follow-up, two patients had a score of 3 and one was a 6.
Dr. Nguyen’s presentation met with undisguised audience skepticism.
“What is the theoretical basis to treat very severe epileptic seizures with another epileptic seizure? I mean, what made you do this?” one neurologist asked.
Dr. Nguyen replied that the mechanism of benefit isn’t clear. One possibility is enhanced inhibition of gamma-aminobutyric acid.
“When I describe ECT to the family, the way I think of it is as a hard reset. We tried burst suppression. This is an alternative approach,” she explained.
Another audience member said the treatment strategy smacks of homeopathy.
“These patients have a terrible disorder,” said session co-chair Gregory Krauss, MD. “Three of your four patients ultimately did not do very well.
“The question is, what are you really accomplishing in terms of the underlying encephalopathy and irritability that’s causing this? And are the seizures really a primary factor in their outcome? Is this treatment warranted to try to improve their overall outcome?” asked Dr. Krauss, professor of neurology at Johns Hopkins University, Baltimore.
Dr. Nguyen answered that she can’t say if the seizures are the cause or result of the encephalopathy.
“ECT was done well into the patients’ admission. Our difficulty is that while we were able to stop the seizures, unfortunately we weren’t able to go back in time and save the brain that was lost due to the convulsive and nonconvulsive activity,” she said. Dr. Nguyen added that she and her coinvestigators are interested in exploring the possibility that utilizing ECT earlier might abort the super-refractory status epilepticus sooner and thereby result in better outcomes.
Still, she noted, three of the four patients were able to leave the hospital, and the two survivors show improved cognitive abilities at 51 and 100 months of follow-up.
Dr. Nguyen reported having no financial conflicts.
SOURCE: Nguyen MTV et al. IEC 2019, Abstract P031.
BANGKOK –
“These were highly refractory patients and ECT did contribute to their status termination,” she reported at the congress, sponsored by the International League Against Epilepsy.
Dr. Nguyen, a neurologist at Oregon Health and Science University, Portland, presented a single-center retrospective case series composed of four ECT-treated patients with NORSE (new onset refractory status epilepticus), three of whom experienced marked improvement in their seizure activity, including cessation of their status epilepticus, after completing a course of eight or nine ECT sessions.
A four-patient series may not seem to be compelling evidence, but it’s a significant contribution to the sparse literature regarding this off-label usage of ECT. Indeed, the biggest case series to date consists of eight patients treated at Indiana University, five of whom displayed neurotelemetry or clinical evidence of improvement within 24 hours after completing a full course of ECT (J ECT. 2018 Mar;34[1]:e5-e9. doi: 10.1097/YCT.0000000000000450).
And realistically, these small case series are as good as the supporting evidence is likely to get.
“It would be incredibly difficult to perform a randomized trial, and even a case-control study in such refractory patients would be quite difficult,” she observed.
Super-refractory status epilepticus is the term for seizures persisting despite 24 hours of adequate treatment with benzodiazepines, loading doses of antiepileptic drugs, and anesthetic agents for medically-induced coma, or for seizures that resume after withdrawal of general anesthesia. NORSE is defined as refractory status epilepticus without a history of seizures or a readily identifiable etiology. It is often associated with an autoimmune or paraneoplastic encephalitis. Outcomes are generally poor.
The four ECT-treated patients in the Oregon series were aged 27-48 years. Two had a prodromal viral illness, and a third had a vague prodrome but a negative infectious disease workup. Three patients were on four antiepileptic drugs at the time they began ECT, one was on seven. Two patients had generalized seizures, while the other two had both generalized and focal seizures. Two had normal brain MRI scans and the other two had abnormal MRIs. The patients had CSF white blood cell counts of 2, 5, 10, and 58 per mm3. All patients were on immunotherapy with intravenous corticosteroids, and three of the four were on additional immunomodulatory drugs.
ECT was started on a compassionate-use basis 16-49 days after hospital admission. These were patients who had run out of options, according to Dr. Nguyen.
Three of the four patients returned to consciousness after completing their course of ECT and withdrawal of their general anesthesia, with attenuation of their seizure activity and cessation of their super-refractory status epilepticus. Two of the three were discharged to a rehabilitation facility, including one who eventually could ambulate with slight assistance. Another patient died after discharge, while the fourth patient died during the initial hospital stay due to septic shock unrelated to the ECT.
At discharge, one patient had a modified Rankin Scale score (mRS) of 4, and two had a score of 5. At follow-up, two patients had a score of 3 and one was a 6.
Dr. Nguyen’s presentation met with undisguised audience skepticism.
“What is the theoretical basis to treat very severe epileptic seizures with another epileptic seizure? I mean, what made you do this?” one neurologist asked.
Dr. Nguyen replied that the mechanism of benefit isn’t clear. One possibility is enhanced inhibition of gamma-aminobutyric acid.
“When I describe ECT to the family, the way I think of it is as a hard reset. We tried burst suppression. This is an alternative approach,” she explained.
Another audience member said the treatment strategy smacks of homeopathy.
“These patients have a terrible disorder,” said session co-chair Gregory Krauss, MD. “Three of your four patients ultimately did not do very well.
“The question is, what are you really accomplishing in terms of the underlying encephalopathy and irritability that’s causing this? And are the seizures really a primary factor in their outcome? Is this treatment warranted to try to improve their overall outcome?” asked Dr. Krauss, professor of neurology at Johns Hopkins University, Baltimore.
Dr. Nguyen answered that she can’t say if the seizures are the cause or result of the encephalopathy.
“ECT was done well into the patients’ admission. Our difficulty is that while we were able to stop the seizures, unfortunately we weren’t able to go back in time and save the brain that was lost due to the convulsive and nonconvulsive activity,” she said. Dr. Nguyen added that she and her coinvestigators are interested in exploring the possibility that utilizing ECT earlier might abort the super-refractory status epilepticus sooner and thereby result in better outcomes.
Still, she noted, three of the four patients were able to leave the hospital, and the two survivors show improved cognitive abilities at 51 and 100 months of follow-up.
Dr. Nguyen reported having no financial conflicts.
SOURCE: Nguyen MTV et al. IEC 2019, Abstract P031.
BANGKOK –
“These were highly refractory patients and ECT did contribute to their status termination,” she reported at the congress, sponsored by the International League Against Epilepsy.
Dr. Nguyen, a neurologist at Oregon Health and Science University, Portland, presented a single-center retrospective case series composed of four ECT-treated patients with NORSE (new onset refractory status epilepticus), three of whom experienced marked improvement in their seizure activity, including cessation of their status epilepticus, after completing a course of eight or nine ECT sessions.
A four-patient series may not seem to be compelling evidence, but it’s a significant contribution to the sparse literature regarding this off-label usage of ECT. Indeed, the biggest case series to date consists of eight patients treated at Indiana University, five of whom displayed neurotelemetry or clinical evidence of improvement within 24 hours after completing a full course of ECT (J ECT. 2018 Mar;34[1]:e5-e9. doi: 10.1097/YCT.0000000000000450).
And realistically, these small case series are as good as the supporting evidence is likely to get.
“It would be incredibly difficult to perform a randomized trial, and even a case-control study in such refractory patients would be quite difficult,” she observed.
Super-refractory status epilepticus is the term for seizures persisting despite 24 hours of adequate treatment with benzodiazepines, loading doses of antiepileptic drugs, and anesthetic agents for medically-induced coma, or for seizures that resume after withdrawal of general anesthesia. NORSE is defined as refractory status epilepticus without a history of seizures or a readily identifiable etiology. It is often associated with an autoimmune or paraneoplastic encephalitis. Outcomes are generally poor.
The four ECT-treated patients in the Oregon series were aged 27-48 years. Two had a prodromal viral illness, and a third had a vague prodrome but a negative infectious disease workup. Three patients were on four antiepileptic drugs at the time they began ECT, one was on seven. Two patients had generalized seizures, while the other two had both generalized and focal seizures. Two had normal brain MRI scans and the other two had abnormal MRIs. The patients had CSF white blood cell counts of 2, 5, 10, and 58 per mm3. All patients were on immunotherapy with intravenous corticosteroids, and three of the four were on additional immunomodulatory drugs.
ECT was started on a compassionate-use basis 16-49 days after hospital admission. These were patients who had run out of options, according to Dr. Nguyen.
Three of the four patients returned to consciousness after completing their course of ECT and withdrawal of their general anesthesia, with attenuation of their seizure activity and cessation of their super-refractory status epilepticus. Two of the three were discharged to a rehabilitation facility, including one who eventually could ambulate with slight assistance. Another patient died after discharge, while the fourth patient died during the initial hospital stay due to septic shock unrelated to the ECT.
At discharge, one patient had a modified Rankin Scale score (mRS) of 4, and two had a score of 5. At follow-up, two patients had a score of 3 and one was a 6.
Dr. Nguyen’s presentation met with undisguised audience skepticism.
“What is the theoretical basis to treat very severe epileptic seizures with another epileptic seizure? I mean, what made you do this?” one neurologist asked.
Dr. Nguyen replied that the mechanism of benefit isn’t clear. One possibility is enhanced inhibition of gamma-aminobutyric acid.
“When I describe ECT to the family, the way I think of it is as a hard reset. We tried burst suppression. This is an alternative approach,” she explained.
Another audience member said the treatment strategy smacks of homeopathy.
“These patients have a terrible disorder,” said session co-chair Gregory Krauss, MD. “Three of your four patients ultimately did not do very well.
“The question is, what are you really accomplishing in terms of the underlying encephalopathy and irritability that’s causing this? And are the seizures really a primary factor in their outcome? Is this treatment warranted to try to improve their overall outcome?” asked Dr. Krauss, professor of neurology at Johns Hopkins University, Baltimore.
Dr. Nguyen answered that she can’t say if the seizures are the cause or result of the encephalopathy.
“ECT was done well into the patients’ admission. Our difficulty is that while we were able to stop the seizures, unfortunately we weren’t able to go back in time and save the brain that was lost due to the convulsive and nonconvulsive activity,” she said. Dr. Nguyen added that she and her coinvestigators are interested in exploring the possibility that utilizing ECT earlier might abort the super-refractory status epilepticus sooner and thereby result in better outcomes.
Still, she noted, three of the four patients were able to leave the hospital, and the two survivors show improved cognitive abilities at 51 and 100 months of follow-up.
Dr. Nguyen reported having no financial conflicts.
SOURCE: Nguyen MTV et al. IEC 2019, Abstract P031.
REPORTING FROM IEC 2019
Novel score spots high-risk febrile children in ED
LJUBLJANA, SLOVENIA – A new age-adjusted quick Sequential Organ Failure Assessment (qSOFA) score designed for use in children presenting to the ED with fever showed good predictive value for admission to critical care within the next 48 hours, Aakash Khanijau, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.

“In the needle-in-a-haystack scenario that’s seen in pediatric emergency departments, our novel, age-adjusted qSOFA score could potentially improve the rapid identification and treatment of children with suspected sepsis presenting to the ED,” said Dr. Khanijau of the University of Liverpool (England).
He presented an exceptionally large retrospective validation study of the score’s performance in 12,393 children (median age, 2.5 years) who presented to EDs with fever, of whom 1,521 were admitted for suspected sepsis. Of the hospitalized children, 145 were admitted to critical care within the first 48 hours.
The pediatric qSOFA score had 72% sensitivity and 85% specificity for critical care admission within 48 hours, with a positive predictive value of 5.4% and, more importantly, a whopping negative predictive value of 99.6%.
“That very high negative predictive value underlines the powerful discriminatory nature of our tool in the emergency department setting,” Dr. Khanijau observed, adding that the score’s area under the receiver operating characteristic curve was 0.81, which is considered a good predictive value.
The impetus for developing an age-adjusted pediatric qSOFA score stems from the fact that the original qSOFA score was designed for rapid assessment of adults with suspected sepsis and isn’t applicable in children. Other existing scores, including SIRS (the Systemic Inflammatory Response Syndrome criteria), the full SOFA, and PELOD-2 (the Pediatric Logistic Organ Dysfunction score), take longer to determine than the adapted qSOFA in a setting where speed is of the essence, he explained.
The original qSOFA components are altered mentation, systolic blood pressure, and respiratory rate. The novel score developed by Dr. Khanijau and coworkers swaps out systolic BP in favor of capillary refill time and age-adjusted heart rate using the thresholds previously established in a landmark study from the Children’s Hospital of Philadelphia (Pediatrics. 2013 Apr;131[4]:e1150-7.)
“Our reasoning here is that arterial hypertension is known to be a much later sign of circulatory compromise in children and may provide less discriminatory value than signs such as delayed capillary refill time and tachycardia early in presentation in the emergency department,” according to Dr. Khanijau.
The novel scoring system features four criteria. One point each is given for a capillary refill time of 3 seconds or longer; anything less than “Alert” on the Alert, Responds to Voice, Respond to Pain, and Unresponsive scale; a heart rate above the 99th percentile on the age-adjusted curves; and a respiratory rate above the age-adjusted 99th percentile. Thus, scores can range from 0 to 4. In the validation study, a score of 2 or more spelled a 890% increased likelihood of being admitted to a critical care setting within 48 hours. It was also associated with a 100-fold increased likelihood of death during the hospitalization, which occurred in 10 children.
Asked how the new predictive score could change clinical management, Dr. Khanijau replied, “I think the key thing it does here is it identifies the children at risk of requiring critical care and should therefore motivate us in the children achieving that threshold to promptly investigate thoroughly for suspected sepsis using the more comprehensive tools, like the full SOFA.”
He reported having no financial conflicts of interest regarding his study.
SOURCE: Khanijau A et al. ESPID 2019, Abstract.
LJUBLJANA, SLOVENIA – A new age-adjusted quick Sequential Organ Failure Assessment (qSOFA) score designed for use in children presenting to the ED with fever showed good predictive value for admission to critical care within the next 48 hours, Aakash Khanijau, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
“In the needle-in-a-haystack scenario that’s seen in pediatric emergency departments, our novel, age-adjusted qSOFA score could potentially improve the rapid identification and treatment of children with suspected sepsis presenting to the ED,” said Dr. Khanijau of the University of Liverpool (England).
He presented an exceptionally large retrospective validation study of the score’s performance in 12,393 children (median age, 2.5 years) who presented to EDs with fever, of whom 1,521 were admitted for suspected sepsis. Of the hospitalized children, 145 were admitted to critical care within the first 48 hours.
The pediatric qSOFA score had 72% sensitivity and 85% specificity for critical care admission within 48 hours, with a positive predictive value of 5.4% and, more importantly, a whopping negative predictive value of 99.6%.
“That very high negative predictive value underlines the powerful discriminatory nature of our tool in the emergency department setting,” Dr. Khanijau observed, adding that the score’s area under the receiver operating characteristic curve was 0.81, which is considered a good predictive value.
The impetus for developing an age-adjusted pediatric qSOFA score stems from the fact that the original qSOFA score was designed for rapid assessment of adults with suspected sepsis and isn’t applicable in children. Other existing scores, including SIRS (the Systemic Inflammatory Response Syndrome criteria), the full SOFA, and PELOD-2 (the Pediatric Logistic Organ Dysfunction score), take longer to determine than the adapted qSOFA in a setting where speed is of the essence, he explained.
The original qSOFA components are altered mentation, systolic blood pressure, and respiratory rate. The novel score developed by Dr. Khanijau and coworkers swaps out systolic BP in favor of capillary refill time and age-adjusted heart rate using the thresholds previously established in a landmark study from the Children’s Hospital of Philadelphia (Pediatrics. 2013 Apr;131[4]:e1150-7.)
“Our reasoning here is that arterial hypertension is known to be a much later sign of circulatory compromise in children and may provide less discriminatory value than signs such as delayed capillary refill time and tachycardia early in presentation in the emergency department,” according to Dr. Khanijau.
The novel scoring system features four criteria. One point each is given for a capillary refill time of 3 seconds or longer; anything less than “Alert” on the Alert, Responds to Voice, Respond to Pain, and Unresponsive scale; a heart rate above the 99th percentile on the age-adjusted curves; and a respiratory rate above the age-adjusted 99th percentile. Thus, scores can range from 0 to 4. In the validation study, a score of 2 or more spelled a 890% increased likelihood of being admitted to a critical care setting within 48 hours. It was also associated with a 100-fold increased likelihood of death during the hospitalization, which occurred in 10 children.
Asked how the new predictive score could change clinical management, Dr. Khanijau replied, “I think the key thing it does here is it identifies the children at risk of requiring critical care and should therefore motivate us in the children achieving that threshold to promptly investigate thoroughly for suspected sepsis using the more comprehensive tools, like the full SOFA.”
He reported having no financial conflicts of interest regarding his study.
SOURCE: Khanijau A et al. ESPID 2019, Abstract.
LJUBLJANA, SLOVENIA – A new age-adjusted quick Sequential Organ Failure Assessment (qSOFA) score designed for use in children presenting to the ED with fever showed good predictive value for admission to critical care within the next 48 hours, Aakash Khanijau, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
“In the needle-in-a-haystack scenario that’s seen in pediatric emergency departments, our novel, age-adjusted qSOFA score could potentially improve the rapid identification and treatment of children with suspected sepsis presenting to the ED,” said Dr. Khanijau of the University of Liverpool (England).
He presented an exceptionally large retrospective validation study of the score’s performance in 12,393 children (median age, 2.5 years) who presented to EDs with fever, of whom 1,521 were admitted for suspected sepsis. Of the hospitalized children, 145 were admitted to critical care within the first 48 hours.
The pediatric qSOFA score had 72% sensitivity and 85% specificity for critical care admission within 48 hours, with a positive predictive value of 5.4% and, more importantly, a whopping negative predictive value of 99.6%.
“That very high negative predictive value underlines the powerful discriminatory nature of our tool in the emergency department setting,” Dr. Khanijau observed, adding that the score’s area under the receiver operating characteristic curve was 0.81, which is considered a good predictive value.
The impetus for developing an age-adjusted pediatric qSOFA score stems from the fact that the original qSOFA score was designed for rapid assessment of adults with suspected sepsis and isn’t applicable in children. Other existing scores, including SIRS (the Systemic Inflammatory Response Syndrome criteria), the full SOFA, and PELOD-2 (the Pediatric Logistic Organ Dysfunction score), take longer to determine than the adapted qSOFA in a setting where speed is of the essence, he explained.
The original qSOFA components are altered mentation, systolic blood pressure, and respiratory rate. The novel score developed by Dr. Khanijau and coworkers swaps out systolic BP in favor of capillary refill time and age-adjusted heart rate using the thresholds previously established in a landmark study from the Children’s Hospital of Philadelphia (Pediatrics. 2013 Apr;131[4]:e1150-7.)
“Our reasoning here is that arterial hypertension is known to be a much later sign of circulatory compromise in children and may provide less discriminatory value than signs such as delayed capillary refill time and tachycardia early in presentation in the emergency department,” according to Dr. Khanijau.
The novel scoring system features four criteria. One point each is given for a capillary refill time of 3 seconds or longer; anything less than “Alert” on the Alert, Responds to Voice, Respond to Pain, and Unresponsive scale; a heart rate above the 99th percentile on the age-adjusted curves; and a respiratory rate above the age-adjusted 99th percentile. Thus, scores can range from 0 to 4. In the validation study, a score of 2 or more spelled a 890% increased likelihood of being admitted to a critical care setting within 48 hours. It was also associated with a 100-fold increased likelihood of death during the hospitalization, which occurred in 10 children.
Asked how the new predictive score could change clinical management, Dr. Khanijau replied, “I think the key thing it does here is it identifies the children at risk of requiring critical care and should therefore motivate us in the children achieving that threshold to promptly investigate thoroughly for suspected sepsis using the more comprehensive tools, like the full SOFA.”
He reported having no financial conflicts of interest regarding his study.
SOURCE: Khanijau A et al. ESPID 2019, Abstract.
REPORTING FROM ESPID 2019
Procalcitonin advocated to help rule out bacterial infections
SEATTLE – Procalcitonin, a marker of bacterial infection, rises and peaks sooner than C-reactive protein (CRP), and is especially useful to help rule out invasive bacterial infections in young infants and pediatric community acquired pneumonia due to typical bacteria, according to a presentation at the 2019 Pediatric Hospital Medicine Conference.

It’s “excellent for identifying low risk patients” and has the potential to decrease lumbar punctures and antibiotic exposure, but “the specificity isn’t great,” so there’s the potential for false positives, said Russell McCulloh, MD, a pediatric infectious disease specialist at the University of Nebraska Medical Center, Omaha.
There was great interest in procalcitonin at the meeting; the presentation room was packed, with a line out the door. It’s used mostly in Europe at this point. Testing is available in many U.S. hospitals, but a large majority of audience members, when polled, said they don’t currently use it in clinical practice, and that it’s not a part of diagnostic algorithms at their institutions.
Levels of procalcitonin, a calcitonin precursor normally produced by the thyroid, are low or undetectable in healthy people, but inflammation, be it from infectious or noninfectious causes, triggers production by parenchymal cells throughout the body.
Levels began to rise as early as 2.5 hours after healthy subjects in one study were injected with bacterial endotoxins, and peaked as early as 6 hours; CRP, in contrast, started to rise after 12 hours, and peaked at 30 hours. Procalcitonin levels also seem to correlate with bacterial load and severity of infection, said Nivedita Srinivas, MD, a pediatric infectious disease specialist at Stanford (Calif.) University (J Pediatr Intensive Care. 2016 Dec;5[4]:162-71).
Due to time, the presenters focused their talk on community acquired pneumonia (CAP) and invasive bacterial infections (IBI) in young infants, meaning essentially bacteremia and meningitis.
Different studies use different cutoffs, but a procalcitonin below, for instance, 0.5 ng/mL is “certainly more sensitive [for IBI] than any single biomarker we currently use,” including CRP, white blood cells, and absolute neutrophil count (ANC). “If it’s negative, you’re really confident it’s negative,” but “a positive test does not necessarily indicate the presence of IBI,” Dr. McCulloh said (Pediatrics. 2012 Nov;130[5]:815-22).
“Procalcitonin works really well as part of a validated step-wise rule” that includes, for instance, CRP and ANC; “I think that’s where its utility is. On its own, it is not a substitute for you examining the patient and doing your basic risk stratification, but it may enhance your decision making incrementally above what we currently have,” he said.
Meanwhile, in a study of 532 children a median age of 2.4 years with radiographically confirmed CAP, procalcitonin levels were a median of 6.1 ng/mL in children whose pneumonia was caused by Streptococcus pneumoniae or other typical bacteria, and no child infected with typical bacteria had a level under 0.1 ng/mL. Below that level, “you can be very sure you do not have typical bacteria pneumonia,” said Marie Wang, MD, also a pediatric infectious disease specialist at Stanford (J Pediatric Infect Dis Soc. 2018 Feb 19;7[1]:46-53).
As procalcitonin levels went up, the likelihood of having bacterial pneumonia increased; at 2 ng/mL, 26% of subjects were infected with typical bacteria, “but even in that group, 58% still had viral infection, so you are still detecting a lot of viral” disease, she said.
Prolcalcitonin-guided therapy – antibiotics until patients fall below a level of 0.25 ng/ml, for instance – has also been associated with decreased antibiotic exposure (Respir Med. 2011 Dec;105[12]:1939-45).
The speakers had no disclosures. The meeting was sponsored by the Society of Hospital Medicine, the American Academy of Pediatrics, and the Academic Pediatric Association.
SEATTLE – Procalcitonin, a marker of bacterial infection, rises and peaks sooner than C-reactive protein (CRP), and is especially useful to help rule out invasive bacterial infections in young infants and pediatric community acquired pneumonia due to typical bacteria, according to a presentation at the 2019 Pediatric Hospital Medicine Conference.
It’s “excellent for identifying low risk patients” and has the potential to decrease lumbar punctures and antibiotic exposure, but “the specificity isn’t great,” so there’s the potential for false positives, said Russell McCulloh, MD, a pediatric infectious disease specialist at the University of Nebraska Medical Center, Omaha.
There was great interest in procalcitonin at the meeting; the presentation room was packed, with a line out the door. It’s used mostly in Europe at this point. Testing is available in many U.S. hospitals, but a large majority of audience members, when polled, said they don’t currently use it in clinical practice, and that it’s not a part of diagnostic algorithms at their institutions.
Levels of procalcitonin, a calcitonin precursor normally produced by the thyroid, are low or undetectable in healthy people, but inflammation, be it from infectious or noninfectious causes, triggers production by parenchymal cells throughout the body.
Levels began to rise as early as 2.5 hours after healthy subjects in one study were injected with bacterial endotoxins, and peaked as early as 6 hours; CRP, in contrast, started to rise after 12 hours, and peaked at 30 hours. Procalcitonin levels also seem to correlate with bacterial load and severity of infection, said Nivedita Srinivas, MD, a pediatric infectious disease specialist at Stanford (Calif.) University (J Pediatr Intensive Care. 2016 Dec;5[4]:162-71).
Due to time, the presenters focused their talk on community acquired pneumonia (CAP) and invasive bacterial infections (IBI) in young infants, meaning essentially bacteremia and meningitis.
Different studies use different cutoffs, but a procalcitonin below, for instance, 0.5 ng/mL is “certainly more sensitive [for IBI] than any single biomarker we currently use,” including CRP, white blood cells, and absolute neutrophil count (ANC). “If it’s negative, you’re really confident it’s negative,” but “a positive test does not necessarily indicate the presence of IBI,” Dr. McCulloh said (Pediatrics. 2012 Nov;130[5]:815-22).
“Procalcitonin works really well as part of a validated step-wise rule” that includes, for instance, CRP and ANC; “I think that’s where its utility is. On its own, it is not a substitute for you examining the patient and doing your basic risk stratification, but it may enhance your decision making incrementally above what we currently have,” he said.
Meanwhile, in a study of 532 children a median age of 2.4 years with radiographically confirmed CAP, procalcitonin levels were a median of 6.1 ng/mL in children whose pneumonia was caused by Streptococcus pneumoniae or other typical bacteria, and no child infected with typical bacteria had a level under 0.1 ng/mL. Below that level, “you can be very sure you do not have typical bacteria pneumonia,” said Marie Wang, MD, also a pediatric infectious disease specialist at Stanford (J Pediatric Infect Dis Soc. 2018 Feb 19;7[1]:46-53).
As procalcitonin levels went up, the likelihood of having bacterial pneumonia increased; at 2 ng/mL, 26% of subjects were infected with typical bacteria, “but even in that group, 58% still had viral infection, so you are still detecting a lot of viral” disease, she said.
Prolcalcitonin-guided therapy – antibiotics until patients fall below a level of 0.25 ng/ml, for instance – has also been associated with decreased antibiotic exposure (Respir Med. 2011 Dec;105[12]:1939-45).
The speakers had no disclosures. The meeting was sponsored by the Society of Hospital Medicine, the American Academy of Pediatrics, and the Academic Pediatric Association.
SEATTLE – Procalcitonin, a marker of bacterial infection, rises and peaks sooner than C-reactive protein (CRP), and is especially useful to help rule out invasive bacterial infections in young infants and pediatric community acquired pneumonia due to typical bacteria, according to a presentation at the 2019 Pediatric Hospital Medicine Conference.
It’s “excellent for identifying low risk patients” and has the potential to decrease lumbar punctures and antibiotic exposure, but “the specificity isn’t great,” so there’s the potential for false positives, said Russell McCulloh, MD, a pediatric infectious disease specialist at the University of Nebraska Medical Center, Omaha.
There was great interest in procalcitonin at the meeting; the presentation room was packed, with a line out the door. It’s used mostly in Europe at this point. Testing is available in many U.S. hospitals, but a large majority of audience members, when polled, said they don’t currently use it in clinical practice, and that it’s not a part of diagnostic algorithms at their institutions.
Levels of procalcitonin, a calcitonin precursor normally produced by the thyroid, are low or undetectable in healthy people, but inflammation, be it from infectious or noninfectious causes, triggers production by parenchymal cells throughout the body.
Levels began to rise as early as 2.5 hours after healthy subjects in one study were injected with bacterial endotoxins, and peaked as early as 6 hours; CRP, in contrast, started to rise after 12 hours, and peaked at 30 hours. Procalcitonin levels also seem to correlate with bacterial load and severity of infection, said Nivedita Srinivas, MD, a pediatric infectious disease specialist at Stanford (Calif.) University (J Pediatr Intensive Care. 2016 Dec;5[4]:162-71).
Due to time, the presenters focused their talk on community acquired pneumonia (CAP) and invasive bacterial infections (IBI) in young infants, meaning essentially bacteremia and meningitis.
Different studies use different cutoffs, but a procalcitonin below, for instance, 0.5 ng/mL is “certainly more sensitive [for IBI] than any single biomarker we currently use,” including CRP, white blood cells, and absolute neutrophil count (ANC). “If it’s negative, you’re really confident it’s negative,” but “a positive test does not necessarily indicate the presence of IBI,” Dr. McCulloh said (Pediatrics. 2012 Nov;130[5]:815-22).
“Procalcitonin works really well as part of a validated step-wise rule” that includes, for instance, CRP and ANC; “I think that’s where its utility is. On its own, it is not a substitute for you examining the patient and doing your basic risk stratification, but it may enhance your decision making incrementally above what we currently have,” he said.
Meanwhile, in a study of 532 children a median age of 2.4 years with radiographically confirmed CAP, procalcitonin levels were a median of 6.1 ng/mL in children whose pneumonia was caused by Streptococcus pneumoniae or other typical bacteria, and no child infected with typical bacteria had a level under 0.1 ng/mL. Below that level, “you can be very sure you do not have typical bacteria pneumonia,” said Marie Wang, MD, also a pediatric infectious disease specialist at Stanford (J Pediatric Infect Dis Soc. 2018 Feb 19;7[1]:46-53).
As procalcitonin levels went up, the likelihood of having bacterial pneumonia increased; at 2 ng/mL, 26% of subjects were infected with typical bacteria, “but even in that group, 58% still had viral infection, so you are still detecting a lot of viral” disease, she said.
Prolcalcitonin-guided therapy – antibiotics until patients fall below a level of 0.25 ng/ml, for instance – has also been associated with decreased antibiotic exposure (Respir Med. 2011 Dec;105[12]:1939-45).
The speakers had no disclosures. The meeting was sponsored by the Society of Hospital Medicine, the American Academy of Pediatrics, and the Academic Pediatric Association.
EXPERT ANALYSIS FROM PHM 2019
Center’s experience casts doubt on clinical utility of NGS
Next-generation sequencing (NGS) of tumor samples seldom changes patient management, and even when it does prompt off-label therapy, outcomes are usually poor, one center’s experience suggests.
“NGS has allowed more personalized medicine in oncology. It is well established that treatment of certain actionable mutations improves outcomes in many cancer types,” wrote Gregory J. Kubicek, MD, of MD Anderson Cancer Center at Cooper in Camden, N.J., and colleagues.
However, evidence of its utility to date has been mixed, and key trials – NCI-MPACT (National Cancer Institute Molecular Profiling–Based Assignment of Cancer Therapy) and NCI-MATCH (National Cancer Institute Molecular Analysis for Therapy Choice)—are still ongoing. “In the interim, the oncologist must make clinical decisions with limited empiric data but an exponentially increasing number of options,” they noted.
The investigators studied outcomes of the first 305 consecutive patients at their institution for whom tissue samples were sent to FoundationOne for NGS testing between March 2014 and April 2017. On average, the patients had received two lines of therapy, and the test was ordered 1.1 years from diagnosis of metastatic disease.
Study findings reported in the Journal of Oncology Practice showed that 116 of the tests were unusable because they did not yield a report (most often as a result of insufficient tissue) or yielded a report that could not be acted on owing to follow-up issues (patient loss of contact, transfer to hospice, or death).
Of the 189 potentially usable tests, 40.2% and 66.7% showed an aberration targetable by on-label therapies and off-label therapies, respectively. And fully 89.9% had actionable aberrations via all potential avenues, including clinical trials.
However, only 11.1% of the 189 potentially usable tests (and merely 8.3% of 253 completed tests and 6.9% of all 305 ordered tests) yielded a change in management, including use of on-label or off-label therapies, enrollment in clinical trials, or discontinuation of medications with a predicted poor response.
Of the six patients who were started on an off-label therapy, the median duration of treatment was 46 days, with half of these patients each stopping therapy because of death or because of progression.
“A vast majority of NGS assay results were not actively incorporated into clinical decision making, despite many assays indicating potential on- or off-label therapies,” Dr. Kubicek and coinvestigators wrote. “Given the escalating cost of medical care and scrutiny thereof, it is important to analyze whether tests are changing management and order tests appropriately.”
Several factors may explain the observed low use of NGS test results, they noted. For example, many patients were heavily pretreated, so some NGS-detected mutations would have already been known. Also, clinicians at the center had little experience with NGS testing.
“A variety of factors make precisely defining the utility of these assays in clinical decision making difficult, but we can certainly conclude that we have observed substantial costs with few discernible benefits,” the investigators stated. “It is possible that there will be greater use in the future as familiarity with these assays increases. Similarly, although we found poor outcomes with NGS-directed off-label therapies, we will eagerly await the results of NCI-MPACT and NCI-MATCH.”
Dr. Kubicek disclosed no relevant conflicts of interest. The study did not receive any specific funding.
SOURCE: Davis W et al. J Oncol Pract. 2019 Aug 2. doi: 10.1200/JOP.19.00269.
Next-generation sequencing (NGS) of tumor samples seldom changes patient management, and even when it does prompt off-label therapy, outcomes are usually poor, one center’s experience suggests.
“NGS has allowed more personalized medicine in oncology. It is well established that treatment of certain actionable mutations improves outcomes in many cancer types,” wrote Gregory J. Kubicek, MD, of MD Anderson Cancer Center at Cooper in Camden, N.J., and colleagues.
However, evidence of its utility to date has been mixed, and key trials – NCI-MPACT (National Cancer Institute Molecular Profiling–Based Assignment of Cancer Therapy) and NCI-MATCH (National Cancer Institute Molecular Analysis for Therapy Choice)—are still ongoing. “In the interim, the oncologist must make clinical decisions with limited empiric data but an exponentially increasing number of options,” they noted.
The investigators studied outcomes of the first 305 consecutive patients at their institution for whom tissue samples were sent to FoundationOne for NGS testing between March 2014 and April 2017. On average, the patients had received two lines of therapy, and the test was ordered 1.1 years from diagnosis of metastatic disease.
Study findings reported in the Journal of Oncology Practice showed that 116 of the tests were unusable because they did not yield a report (most often as a result of insufficient tissue) or yielded a report that could not be acted on owing to follow-up issues (patient loss of contact, transfer to hospice, or death).
Of the 189 potentially usable tests, 40.2% and 66.7% showed an aberration targetable by on-label therapies and off-label therapies, respectively. And fully 89.9% had actionable aberrations via all potential avenues, including clinical trials.
However, only 11.1% of the 189 potentially usable tests (and merely 8.3% of 253 completed tests and 6.9% of all 305 ordered tests) yielded a change in management, including use of on-label or off-label therapies, enrollment in clinical trials, or discontinuation of medications with a predicted poor response.
Of the six patients who were started on an off-label therapy, the median duration of treatment was 46 days, with half of these patients each stopping therapy because of death or because of progression.
“A vast majority of NGS assay results were not actively incorporated into clinical decision making, despite many assays indicating potential on- or off-label therapies,” Dr. Kubicek and coinvestigators wrote. “Given the escalating cost of medical care and scrutiny thereof, it is important to analyze whether tests are changing management and order tests appropriately.”
Several factors may explain the observed low use of NGS test results, they noted. For example, many patients were heavily pretreated, so some NGS-detected mutations would have already been known. Also, clinicians at the center had little experience with NGS testing.
“A variety of factors make precisely defining the utility of these assays in clinical decision making difficult, but we can certainly conclude that we have observed substantial costs with few discernible benefits,” the investigators stated. “It is possible that there will be greater use in the future as familiarity with these assays increases. Similarly, although we found poor outcomes with NGS-directed off-label therapies, we will eagerly await the results of NCI-MPACT and NCI-MATCH.”
Dr. Kubicek disclosed no relevant conflicts of interest. The study did not receive any specific funding.
SOURCE: Davis W et al. J Oncol Pract. 2019 Aug 2. doi: 10.1200/JOP.19.00269.
Next-generation sequencing (NGS) of tumor samples seldom changes patient management, and even when it does prompt off-label therapy, outcomes are usually poor, one center’s experience suggests.
“NGS has allowed more personalized medicine in oncology. It is well established that treatment of certain actionable mutations improves outcomes in many cancer types,” wrote Gregory J. Kubicek, MD, of MD Anderson Cancer Center at Cooper in Camden, N.J., and colleagues.
However, evidence of its utility to date has been mixed, and key trials – NCI-MPACT (National Cancer Institute Molecular Profiling–Based Assignment of Cancer Therapy) and NCI-MATCH (National Cancer Institute Molecular Analysis for Therapy Choice)—are still ongoing. “In the interim, the oncologist must make clinical decisions with limited empiric data but an exponentially increasing number of options,” they noted.
The investigators studied outcomes of the first 305 consecutive patients at their institution for whom tissue samples were sent to FoundationOne for NGS testing between March 2014 and April 2017. On average, the patients had received two lines of therapy, and the test was ordered 1.1 years from diagnosis of metastatic disease.
Study findings reported in the Journal of Oncology Practice showed that 116 of the tests were unusable because they did not yield a report (most often as a result of insufficient tissue) or yielded a report that could not be acted on owing to follow-up issues (patient loss of contact, transfer to hospice, or death).
Of the 189 potentially usable tests, 40.2% and 66.7% showed an aberration targetable by on-label therapies and off-label therapies, respectively. And fully 89.9% had actionable aberrations via all potential avenues, including clinical trials.
However, only 11.1% of the 189 potentially usable tests (and merely 8.3% of 253 completed tests and 6.9% of all 305 ordered tests) yielded a change in management, including use of on-label or off-label therapies, enrollment in clinical trials, or discontinuation of medications with a predicted poor response.
Of the six patients who were started on an off-label therapy, the median duration of treatment was 46 days, with half of these patients each stopping therapy because of death or because of progression.
“A vast majority of NGS assay results were not actively incorporated into clinical decision making, despite many assays indicating potential on- or off-label therapies,” Dr. Kubicek and coinvestigators wrote. “Given the escalating cost of medical care and scrutiny thereof, it is important to analyze whether tests are changing management and order tests appropriately.”
Several factors may explain the observed low use of NGS test results, they noted. For example, many patients were heavily pretreated, so some NGS-detected mutations would have already been known. Also, clinicians at the center had little experience with NGS testing.
“A variety of factors make precisely defining the utility of these assays in clinical decision making difficult, but we can certainly conclude that we have observed substantial costs with few discernible benefits,” the investigators stated. “It is possible that there will be greater use in the future as familiarity with these assays increases. Similarly, although we found poor outcomes with NGS-directed off-label therapies, we will eagerly await the results of NCI-MPACT and NCI-MATCH.”
Dr. Kubicek disclosed no relevant conflicts of interest. The study did not receive any specific funding.
SOURCE: Davis W et al. J Oncol Pract. 2019 Aug 2. doi: 10.1200/JOP.19.00269.
FROM THE JOURNAL OF ONCOLOGY PRACTICE
Interview with Andrew Pachner, MD, about the molecular processes of multiple sclerosis
Andrew R. Pachner, MD is the Murray B. Bornstein professor of neurology at Geisel School of Medicine at Dartmouth and director of the Multiple Sclerosis Center at Dartmouth-Hitchcock Medical Center. We spoke to Dr. Pachner about his research into the molecular processes of multiple sclerosis (MS) and the potential impact on patient management.

What do we know about the molecular processes behind relapsing-remitting and progressive MS?
DR. PACHNER: The progress--in terms of molecules--has not been rapid in the field of MS. The only molecular biomarker we use in practice is oligoclonal bands or other measures of immunoglobulin production in the nervous system, and that biomarker was described in 1942. So, it has been a long time since we have seen a relevant molecule that we can use clinically.
But there has been a lot of progress in the general field of neuroinflammation. MS is one of a large number of diseases that results in neuroinflammation and demyelination.
One thing we have learned over time is that there are many different subtypes of MS. They probably have some shared molecular processes, but they also are likely to have divergent molecular processes.
Over the past 5 to 10 years, researchers have been interested in trying to dissect some of the molecular aspects of MS to identify biomarkers that can, in turn, differentiate subtypes of MS. This will help to identify different ways of treating MS that are optimal for individual patients. It is clear that each patient is quite different and unlikely to be standardized in the way they respond to treatment.
The degree to which relapsing-remitting and progressive MS are differentiated on the molecular level is dependent on how much influence there is of the immune system in the periphery. When MS first starts in a patient, the brain has either no or a very primitive immune system, and then over time it changes, and it becomes much more immune-oriented and populated by immune cells and molecules. So, there’s a trend over time of the central nervous system becoming increasingly populated by immune cells and able to make immune molecules.
What has your recent research on murine models representing these disease patterns shown?
DR. PACHNER: Even though in humans there is a continuum from relapsing remitting to progressive, it is not like they are completely separate. Frequently in the middle of relapsing-remitting disease there is some progression over time.
In mouse models, we like things to be very clear and separate. We try to make things as simple as possible because of the complexity of the nervous and immune systems.
The simple model for the relapsing-remitting disease is experimental autoimmune encephalomyelitis (EAE), the most commonly studied model of neuroinflammation.
For the progressive form of MS, we use the Theiler’s virus model, which is a type of virus called the picornavirus that is injected into the brain of mice resulting in a slowly progressive, chronic viral infection that looks very much like progressive MS.
In EAE, the disease is induced by presenting an antigen to the peripheral immune system, allowing cells from the peripheral immune system to enter into the central nervous system. It is a manifestation of inflammation and the immune response is in the periphery. In the Theiler’s model, it is a localized process within the central nervous system because the virus is injected directly into the brain.
We found that in EAE the pattern is very much dominated by what happens in the periphery and the injury is very transient. There are cells that enter the nervous system that cause inflammation and damage, but there are also processes that downregulate those cells and processes and eventually the animal improves--similar to an MS attack.
By contrast, in the Theiler’s model there is progressive injury that is dominated by two molecular processes in the central nervous system that we do not see in relapsing-remitting MS or in EAE, and that is the activation of Type 1 interferons and also a very pronounced immunoglobulin production along with all the molecules that help support plasma cells making immunoglobulin.
These are two different animal models that provide us insight into how the central nervous system can be injured in the course of neuroinflammation and they look to be very different in how they manifest themselves, both in the periphery and in the central nervous system.
How may these new findings impact the future management and treatment of MS?
DR. PACHNER: When I see a patient with MS, I tell them that we absolutely need to focus on your own disease and how it responds, rather than taking too much guidance from MS as a whole. Because each patient with MS is different.
One of the things that we have tried to do is to identify molecular markers that might help us in management and treatment. As an example, we have learned that some patients who present with their first episode of MS do very poorly. These patients have many more attacks and/or have very aggressive progression in terms of their disability so that they potentially could be in a wheelchair within a few years. Other patients have what we call a benign variant MS. These patients may have an initial episode that is not that different than the other patient, but this type of patient may not have anything else for the rest of their life.
We would like to have some differentiation of those two types of patients. In the first example you can try to be very aggressive and minimize the neuroinflammation with powerful immune-suppressing drugs that have a high risk of causing side effects, such as cancer or opportunistic infections, but on the other hand may have a high benefit in preventing future inflammatory events and progressive injury. But that would not be the correct treatment choice for the second patient example.
It would be nice to tailor treatment to a predictive biomarker. That is something we have been working very hard on. Based on some of the animal models, we have identified a molecular signature of inflammatory MS that is very predictive of future events and we are hoping that that will help us differentiate patients. In other words, not just treat every MS patient the same, but identify whether they need a very powerful immunosuppressant drug, or a mildly immunosuppressant drug, or no treatment at all.
If you have a patient who has one attack and never has any other problem with their MS, then they do not need to be on any treatment. Unfortunately, we do not have predictive value at this point for any molecule or any other attribute of the patient at this point in time. We are trying to remedy that.
That is one very practical aspect of our work in trying to understand the biology of the disease better--identifying molecules that are associated with future damage and inflammation and using those in a predictive manner in patients to guide treatment.
Another important aspect is the attempt to understand the biology of neuroinflammation and how it causes both demyelination and progressive injury to neurons.
References:
Pachner AR, DiSano K, Royce DB, Gilli F. Clinical utility of a molecular signature in inflammatory demyelinating diseases. Neurol Neuroimmunol Neuroinflamm.2019;6(1):e520.
Andrew R. Pachner, MD is the Murray B. Bornstein professor of neurology at Geisel School of Medicine at Dartmouth and director of the Multiple Sclerosis Center at Dartmouth-Hitchcock Medical Center. We spoke to Dr. Pachner about his research into the molecular processes of multiple sclerosis (MS) and the potential impact on patient management.
What do we know about the molecular processes behind relapsing-remitting and progressive MS?
DR. PACHNER: The progress--in terms of molecules--has not been rapid in the field of MS. The only molecular biomarker we use in practice is oligoclonal bands or other measures of immunoglobulin production in the nervous system, and that biomarker was described in 1942. So, it has been a long time since we have seen a relevant molecule that we can use clinically.
But there has been a lot of progress in the general field of neuroinflammation. MS is one of a large number of diseases that results in neuroinflammation and demyelination.
One thing we have learned over time is that there are many different subtypes of MS. They probably have some shared molecular processes, but they also are likely to have divergent molecular processes.
Over the past 5 to 10 years, researchers have been interested in trying to dissect some of the molecular aspects of MS to identify biomarkers that can, in turn, differentiate subtypes of MS. This will help to identify different ways of treating MS that are optimal for individual patients. It is clear that each patient is quite different and unlikely to be standardized in the way they respond to treatment.
The degree to which relapsing-remitting and progressive MS are differentiated on the molecular level is dependent on how much influence there is of the immune system in the periphery. When MS first starts in a patient, the brain has either no or a very primitive immune system, and then over time it changes, and it becomes much more immune-oriented and populated by immune cells and molecules. So, there’s a trend over time of the central nervous system becoming increasingly populated by immune cells and able to make immune molecules.
What has your recent research on murine models representing these disease patterns shown?
DR. PACHNER: Even though in humans there is a continuum from relapsing remitting to progressive, it is not like they are completely separate. Frequently in the middle of relapsing-remitting disease there is some progression over time.
In mouse models, we like things to be very clear and separate. We try to make things as simple as possible because of the complexity of the nervous and immune systems.
The simple model for the relapsing-remitting disease is experimental autoimmune encephalomyelitis (EAE), the most commonly studied model of neuroinflammation.
For the progressive form of MS, we use the Theiler’s virus model, which is a type of virus called the picornavirus that is injected into the brain of mice resulting in a slowly progressive, chronic viral infection that looks very much like progressive MS.
In EAE, the disease is induced by presenting an antigen to the peripheral immune system, allowing cells from the peripheral immune system to enter into the central nervous system. It is a manifestation of inflammation and the immune response is in the periphery. In the Theiler’s model, it is a localized process within the central nervous system because the virus is injected directly into the brain.
We found that in EAE the pattern is very much dominated by what happens in the periphery and the injury is very transient. There are cells that enter the nervous system that cause inflammation and damage, but there are also processes that downregulate those cells and processes and eventually the animal improves--similar to an MS attack.
By contrast, in the Theiler’s model there is progressive injury that is dominated by two molecular processes in the central nervous system that we do not see in relapsing-remitting MS or in EAE, and that is the activation of Type 1 interferons and also a very pronounced immunoglobulin production along with all the molecules that help support plasma cells making immunoglobulin.
These are two different animal models that provide us insight into how the central nervous system can be injured in the course of neuroinflammation and they look to be very different in how they manifest themselves, both in the periphery and in the central nervous system.
How may these new findings impact the future management and treatment of MS?
DR. PACHNER: When I see a patient with MS, I tell them that we absolutely need to focus on your own disease and how it responds, rather than taking too much guidance from MS as a whole. Because each patient with MS is different.
One of the things that we have tried to do is to identify molecular markers that might help us in management and treatment. As an example, we have learned that some patients who present with their first episode of MS do very poorly. These patients have many more attacks and/or have very aggressive progression in terms of their disability so that they potentially could be in a wheelchair within a few years. Other patients have what we call a benign variant MS. These patients may have an initial episode that is not that different than the other patient, but this type of patient may not have anything else for the rest of their life.
We would like to have some differentiation of those two types of patients. In the first example you can try to be very aggressive and minimize the neuroinflammation with powerful immune-suppressing drugs that have a high risk of causing side effects, such as cancer or opportunistic infections, but on the other hand may have a high benefit in preventing future inflammatory events and progressive injury. But that would not be the correct treatment choice for the second patient example.
It would be nice to tailor treatment to a predictive biomarker. That is something we have been working very hard on. Based on some of the animal models, we have identified a molecular signature of inflammatory MS that is very predictive of future events and we are hoping that that will help us differentiate patients. In other words, not just treat every MS patient the same, but identify whether they need a very powerful immunosuppressant drug, or a mildly immunosuppressant drug, or no treatment at all.
If you have a patient who has one attack and never has any other problem with their MS, then they do not need to be on any treatment. Unfortunately, we do not have predictive value at this point for any molecule or any other attribute of the patient at this point in time. We are trying to remedy that.
That is one very practical aspect of our work in trying to understand the biology of the disease better--identifying molecules that are associated with future damage and inflammation and using those in a predictive manner in patients to guide treatment.
Another important aspect is the attempt to understand the biology of neuroinflammation and how it causes both demyelination and progressive injury to neurons.
References:
Pachner AR, DiSano K, Royce DB, Gilli F. Clinical utility of a molecular signature in inflammatory demyelinating diseases. Neurol Neuroimmunol Neuroinflamm.2019;6(1):e520.
Andrew R. Pachner, MD is the Murray B. Bornstein professor of neurology at Geisel School of Medicine at Dartmouth and director of the Multiple Sclerosis Center at Dartmouth-Hitchcock Medical Center. We spoke to Dr. Pachner about his research into the molecular processes of multiple sclerosis (MS) and the potential impact on patient management.
What do we know about the molecular processes behind relapsing-remitting and progressive MS?
DR. PACHNER: The progress--in terms of molecules--has not been rapid in the field of MS. The only molecular biomarker we use in practice is oligoclonal bands or other measures of immunoglobulin production in the nervous system, and that biomarker was described in 1942. So, it has been a long time since we have seen a relevant molecule that we can use clinically.
But there has been a lot of progress in the general field of neuroinflammation. MS is one of a large number of diseases that results in neuroinflammation and demyelination.
One thing we have learned over time is that there are many different subtypes of MS. They probably have some shared molecular processes, but they also are likely to have divergent molecular processes.
Over the past 5 to 10 years, researchers have been interested in trying to dissect some of the molecular aspects of MS to identify biomarkers that can, in turn, differentiate subtypes of MS. This will help to identify different ways of treating MS that are optimal for individual patients. It is clear that each patient is quite different and unlikely to be standardized in the way they respond to treatment.
The degree to which relapsing-remitting and progressive MS are differentiated on the molecular level is dependent on how much influence there is of the immune system in the periphery. When MS first starts in a patient, the brain has either no or a very primitive immune system, and then over time it changes, and it becomes much more immune-oriented and populated by immune cells and molecules. So, there’s a trend over time of the central nervous system becoming increasingly populated by immune cells and able to make immune molecules.
What has your recent research on murine models representing these disease patterns shown?
DR. PACHNER: Even though in humans there is a continuum from relapsing remitting to progressive, it is not like they are completely separate. Frequently in the middle of relapsing-remitting disease there is some progression over time.
In mouse models, we like things to be very clear and separate. We try to make things as simple as possible because of the complexity of the nervous and immune systems.
The simple model for the relapsing-remitting disease is experimental autoimmune encephalomyelitis (EAE), the most commonly studied model of neuroinflammation.
For the progressive form of MS, we use the Theiler’s virus model, which is a type of virus called the picornavirus that is injected into the brain of mice resulting in a slowly progressive, chronic viral infection that looks very much like progressive MS.
In EAE, the disease is induced by presenting an antigen to the peripheral immune system, allowing cells from the peripheral immune system to enter into the central nervous system. It is a manifestation of inflammation and the immune response is in the periphery. In the Theiler’s model, it is a localized process within the central nervous system because the virus is injected directly into the brain.
We found that in EAE the pattern is very much dominated by what happens in the periphery and the injury is very transient. There are cells that enter the nervous system that cause inflammation and damage, but there are also processes that downregulate those cells and processes and eventually the animal improves--similar to an MS attack.
By contrast, in the Theiler’s model there is progressive injury that is dominated by two molecular processes in the central nervous system that we do not see in relapsing-remitting MS or in EAE, and that is the activation of Type 1 interferons and also a very pronounced immunoglobulin production along with all the molecules that help support plasma cells making immunoglobulin.
These are two different animal models that provide us insight into how the central nervous system can be injured in the course of neuroinflammation and they look to be very different in how they manifest themselves, both in the periphery and in the central nervous system.
How may these new findings impact the future management and treatment of MS?
DR. PACHNER: When I see a patient with MS, I tell them that we absolutely need to focus on your own disease and how it responds, rather than taking too much guidance from MS as a whole. Because each patient with MS is different.
One of the things that we have tried to do is to identify molecular markers that might help us in management and treatment. As an example, we have learned that some patients who present with their first episode of MS do very poorly. These patients have many more attacks and/or have very aggressive progression in terms of their disability so that they potentially could be in a wheelchair within a few years. Other patients have what we call a benign variant MS. These patients may have an initial episode that is not that different than the other patient, but this type of patient may not have anything else for the rest of their life.
We would like to have some differentiation of those two types of patients. In the first example you can try to be very aggressive and minimize the neuroinflammation with powerful immune-suppressing drugs that have a high risk of causing side effects, such as cancer or opportunistic infections, but on the other hand may have a high benefit in preventing future inflammatory events and progressive injury. But that would not be the correct treatment choice for the second patient example.
It would be nice to tailor treatment to a predictive biomarker. That is something we have been working very hard on. Based on some of the animal models, we have identified a molecular signature of inflammatory MS that is very predictive of future events and we are hoping that that will help us differentiate patients. In other words, not just treat every MS patient the same, but identify whether they need a very powerful immunosuppressant drug, or a mildly immunosuppressant drug, or no treatment at all.
If you have a patient who has one attack and never has any other problem with their MS, then they do not need to be on any treatment. Unfortunately, we do not have predictive value at this point for any molecule or any other attribute of the patient at this point in time. We are trying to remedy that.
That is one very practical aspect of our work in trying to understand the biology of the disease better--identifying molecules that are associated with future damage and inflammation and using those in a predictive manner in patients to guide treatment.
Another important aspect is the attempt to understand the biology of neuroinflammation and how it causes both demyelination and progressive injury to neurons.
References:
Pachner AR, DiSano K, Royce DB, Gilli F. Clinical utility of a molecular signature in inflammatory demyelinating diseases. Neurol Neuroimmunol Neuroinflamm.2019;6(1):e520.

Review reveals lack of data on mild hemophilia A
A literature review has failed to provide new insights regarding the burden of mild hemophilia A.

In the 17 studies reviewed, mean annual bleeding rates (ABRs) were largely unreported. Data on joint pain and damage, quality of life (QOL), societal impacts, and costs of care were limited and inconsistent across the studies.
The review “revealed a lack of evidence” in adults with mild hemophilia A, Flora Peyvandi, MD, PhD, of Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico in Milan and colleagues wrote in Haemophilia.
The researchers reviewed data from 10 studies conducted in Europe, 6 in North America, and 1 in Japan. Six studies were prospective cohort or registry studies, six were retrospective, and five studies were surveys or outcomes research.
The studies included 3,213 patients with mild hemophilia A aged 13 years or older. There were few details on treatment protocols, but patients received factor VIII concentrates, recombinant factor VIII, and desmopressin.
Most studies did not report mean ABRs. For the three that did, the mean ABRs were 0.44, 0.56, and 4.5. Six studies reported the percentage of patients with bleeding events, and those numbers ranged from 5.5% (1/18) to 90.7% (68/75).
Data on joint pain and damage were not standardized across studies, so the researchers were unable to draw any conclusions. One study showed no significant difference in Health Assessment Questionnaire pain score between patients with mild hemophilia A and control subjects. In another study, 5% of patients with mild hemophilia A reported having severe joint pain in the previous year, and 15% of patients reported moderate joint pain.
The researchers also found it difficult to draw conclusions about QOL. Three studies reported QOL data, and all used a different instrument.
In a study using the SF-36, general health and emotional role functioning were both significantly lower for patients with mild hemophilia A than for age-matched healthy control subjects (P less than .05). In a study using the SF-12, the physical component summary was significantly higher for patients with mild hemophilia A than for those with severe disease (P = .014).
In a study using the Haemo-QOL-A, there were no significant differences between patients with mild and severe hemophilia A. However, Dr. Peyvandi and colleagues noted that this study required long-term use of factor VIII concentrate, so the mild hemophilia A patients in this group were “probably not representative” of the overall mild hemophilia A population.
Societal impacts were difficult to assess because of a lack of standardization across studies. One study showed no significant difference in employment between patients with mild hemophilia A and healthy controls. In a U.S.-based study, patients with mild hemophilia A missed an average of 6.2 workdays per year, and 4.7 days were caused by their hemophilia. A study in Italy showed that patients with mild hemophilia A missed an average of 3.4 workdays per year.
Just two studies included data on health care costs for patients with mild hemophilia A. The mean cost of care was €793 per year in a study from Portugal published in 2015. In a U.S. study published in 1995, the annual cost of care was $22,182.
“Considering the limitations of the current body of evidence, higher-quality studies in this area are needed,” Dr. Peyvandi and colleagues wrote. “Such studies would report both bleeding and other clinical outcomes based on common definitions and for a representative population of mild [hemophilia A] adults. Areas for further research include more robust comparison to healthy controls or population norms, especially for QOL and other patient-reported outcomes.”
Seven of the eight researchers reported relationships, including employment, with BioMarin. Dr. Peyvandi reported relationships with Sanofi, Grifols, Novo Nordisk, Roche, Takeda, Sobi, Bioverativ, Spark Therapeutics, Sysmex, and CSL Behring.
SOURCE: Peyvandi F et al. Haemophilia. 2019 Jul 11. doi: 10.1111/hae.13777.
A literature review has failed to provide new insights regarding the burden of mild hemophilia A.
In the 17 studies reviewed, mean annual bleeding rates (ABRs) were largely unreported. Data on joint pain and damage, quality of life (QOL), societal impacts, and costs of care were limited and inconsistent across the studies.
The review “revealed a lack of evidence” in adults with mild hemophilia A, Flora Peyvandi, MD, PhD, of Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico in Milan and colleagues wrote in Haemophilia.
The researchers reviewed data from 10 studies conducted in Europe, 6 in North America, and 1 in Japan. Six studies were prospective cohort or registry studies, six were retrospective, and five studies were surveys or outcomes research.
The studies included 3,213 patients with mild hemophilia A aged 13 years or older. There were few details on treatment protocols, but patients received factor VIII concentrates, recombinant factor VIII, and desmopressin.
Most studies did not report mean ABRs. For the three that did, the mean ABRs were 0.44, 0.56, and 4.5. Six studies reported the percentage of patients with bleeding events, and those numbers ranged from 5.5% (1/18) to 90.7% (68/75).
Data on joint pain and damage were not standardized across studies, so the researchers were unable to draw any conclusions. One study showed no significant difference in Health Assessment Questionnaire pain score between patients with mild hemophilia A and control subjects. In another study, 5% of patients with mild hemophilia A reported having severe joint pain in the previous year, and 15% of patients reported moderate joint pain.
The researchers also found it difficult to draw conclusions about QOL. Three studies reported QOL data, and all used a different instrument.
In a study using the SF-36, general health and emotional role functioning were both significantly lower for patients with mild hemophilia A than for age-matched healthy control subjects (P less than .05). In a study using the SF-12, the physical component summary was significantly higher for patients with mild hemophilia A than for those with severe disease (P = .014).
In a study using the Haemo-QOL-A, there were no significant differences between patients with mild and severe hemophilia A. However, Dr. Peyvandi and colleagues noted that this study required long-term use of factor VIII concentrate, so the mild hemophilia A patients in this group were “probably not representative” of the overall mild hemophilia A population.
Societal impacts were difficult to assess because of a lack of standardization across studies. One study showed no significant difference in employment between patients with mild hemophilia A and healthy controls. In a U.S.-based study, patients with mild hemophilia A missed an average of 6.2 workdays per year, and 4.7 days were caused by their hemophilia. A study in Italy showed that patients with mild hemophilia A missed an average of 3.4 workdays per year.
Just two studies included data on health care costs for patients with mild hemophilia A. The mean cost of care was €793 per year in a study from Portugal published in 2015. In a U.S. study published in 1995, the annual cost of care was $22,182.
“Considering the limitations of the current body of evidence, higher-quality studies in this area are needed,” Dr. Peyvandi and colleagues wrote. “Such studies would report both bleeding and other clinical outcomes based on common definitions and for a representative population of mild [hemophilia A] adults. Areas for further research include more robust comparison to healthy controls or population norms, especially for QOL and other patient-reported outcomes.”
Seven of the eight researchers reported relationships, including employment, with BioMarin. Dr. Peyvandi reported relationships with Sanofi, Grifols, Novo Nordisk, Roche, Takeda, Sobi, Bioverativ, Spark Therapeutics, Sysmex, and CSL Behring.
SOURCE: Peyvandi F et al. Haemophilia. 2019 Jul 11. doi: 10.1111/hae.13777.
A literature review has failed to provide new insights regarding the burden of mild hemophilia A.
In the 17 studies reviewed, mean annual bleeding rates (ABRs) were largely unreported. Data on joint pain and damage, quality of life (QOL), societal impacts, and costs of care were limited and inconsistent across the studies.
The review “revealed a lack of evidence” in adults with mild hemophilia A, Flora Peyvandi, MD, PhD, of Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico in Milan and colleagues wrote in Haemophilia.
The researchers reviewed data from 10 studies conducted in Europe, 6 in North America, and 1 in Japan. Six studies were prospective cohort or registry studies, six were retrospective, and five studies were surveys or outcomes research.
The studies included 3,213 patients with mild hemophilia A aged 13 years or older. There were few details on treatment protocols, but patients received factor VIII concentrates, recombinant factor VIII, and desmopressin.
Most studies did not report mean ABRs. For the three that did, the mean ABRs were 0.44, 0.56, and 4.5. Six studies reported the percentage of patients with bleeding events, and those numbers ranged from 5.5% (1/18) to 90.7% (68/75).
Data on joint pain and damage were not standardized across studies, so the researchers were unable to draw any conclusions. One study showed no significant difference in Health Assessment Questionnaire pain score between patients with mild hemophilia A and control subjects. In another study, 5% of patients with mild hemophilia A reported having severe joint pain in the previous year, and 15% of patients reported moderate joint pain.
The researchers also found it difficult to draw conclusions about QOL. Three studies reported QOL data, and all used a different instrument.
In a study using the SF-36, general health and emotional role functioning were both significantly lower for patients with mild hemophilia A than for age-matched healthy control subjects (P less than .05). In a study using the SF-12, the physical component summary was significantly higher for patients with mild hemophilia A than for those with severe disease (P = .014).
In a study using the Haemo-QOL-A, there were no significant differences between patients with mild and severe hemophilia A. However, Dr. Peyvandi and colleagues noted that this study required long-term use of factor VIII concentrate, so the mild hemophilia A patients in this group were “probably not representative” of the overall mild hemophilia A population.
Societal impacts were difficult to assess because of a lack of standardization across studies. One study showed no significant difference in employment between patients with mild hemophilia A and healthy controls. In a U.S.-based study, patients with mild hemophilia A missed an average of 6.2 workdays per year, and 4.7 days were caused by their hemophilia. A study in Italy showed that patients with mild hemophilia A missed an average of 3.4 workdays per year.
Just two studies included data on health care costs for patients with mild hemophilia A. The mean cost of care was €793 per year in a study from Portugal published in 2015. In a U.S. study published in 1995, the annual cost of care was $22,182.
“Considering the limitations of the current body of evidence, higher-quality studies in this area are needed,” Dr. Peyvandi and colleagues wrote. “Such studies would report both bleeding and other clinical outcomes based on common definitions and for a representative population of mild [hemophilia A] adults. Areas for further research include more robust comparison to healthy controls or population norms, especially for QOL and other patient-reported outcomes.”
Seven of the eight researchers reported relationships, including employment, with BioMarin. Dr. Peyvandi reported relationships with Sanofi, Grifols, Novo Nordisk, Roche, Takeda, Sobi, Bioverativ, Spark Therapeutics, Sysmex, and CSL Behring.
SOURCE: Peyvandi F et al. Haemophilia. 2019 Jul 11. doi: 10.1111/hae.13777.
FROM HAEMOPHILIA
Mortality is high in pediatric superrefractory status epilepticus
BANGKOK – presented by Maggie Lo Yee Yau, MD, at the International Epilepsy Congress.
“Death in these children usually occurred within the first few days after admission to the pediatric ICU,” she said at the congress sponsored by the International League Against Epilepsy.

The study included 15 consecutive patients aged between 1 month and 17 years treated for superrefractory status epilepticus (SRSE) during 2011-2017 at the Chinese University of Hong Kong, where Dr. Yau practices. Seven children died during their index hospital admission, with a median time to death of 8 days. Two more died within several years post discharge.
Morbidity was substantial: At follow-up 1 year after the index episode of SRSE, two patients had a Glasgow Outcome Scale (GOS) score of 3, indicative of severe disability; three patients had moderate disability, with a GOS of 4; and two patients were in a vegetative state, with a GOS of 2, both of whom subsequently died of aspiration pneumonia. Only 1 of the 15 patients had a good recovery. Through 8 years of follow-up, all six survivors had epilepsy. Common nonneurologic deficits included a predisposition to a variety of infections.
By way of background, Dr. Yau noted that convulsive status epilepticus is the most common neurologic emergency in children, with an incidence of about 20 episodes per 100,000. Of affected children, 10%-40% develop refractory status, with reported mortality rates of 16%-43%. SRSE is a term reserved for persistent or recurrent seizures 24 hours or more after onset of general anesthesia for management of refractory status.
The impetus for Dr. Yau’s study was the dearth of data on SRSE in children. The literature consists of a few case series totaling well under 100 patients.
The Hong Kong case series included 15 patients with SRSE who had a median age of 7.9 years, only 1 of whom had preexisting epilepsy, a case of epileptic encephalopathy with severe developmental delay. Of the 15, 12 were boys. The patients were placed on a median of four antiepileptic drugs. Those who survived to discharge spent a median of 17.8 days under general anesthesia and 42.5 days in the pediatric ICU.
The SRSE etiologies included febrile infection–related epilepsy syndrome in two cases, four serious infections, four cases of autoimmune etiology, two cases of epileptic encephalopathy, one patient with hypoxia caused by severe croup, and two of unknown origin despite intensive work-up.
The four in-hospital deaths caused by acute cerebral edema occurred a median 6.5 days after admission. There were also two deaths because of uncontrolled sepsis and one because of intraventricular bleeding secondary to thrombotic thrombocytopenic purpura thought to have occurred as a complication of interactions between the numerous prescribed medications. All six children with an infectious or unknown etiology died in hospital, whereas none of those with an autoimmune etiology, epileptic encephalopathy, or hypoxia did. Duration of anesthesia did not predict mortality.
Other investigators have reported that younger age is associated with higher mortality, but that was not true in the Hong Kong experience. Neither of the two children aged less than 3 years died during their index hospitalization. All 7 deaths occurred in the 13 children age 3 years or older.
When asked whether she thought SRSE or the underlying disorder was the bigger contributor to mortality, Dr. Yau replied that she believes the prolonged refractory seizures may have worsened cerebral edema in some patients and thereby have been the cause of death.
She reported having no financial conflicts regarding her study.
BANGKOK – presented by Maggie Lo Yee Yau, MD, at the International Epilepsy Congress.
“Death in these children usually occurred within the first few days after admission to the pediatric ICU,” she said at the congress sponsored by the International League Against Epilepsy.
The study included 15 consecutive patients aged between 1 month and 17 years treated for superrefractory status epilepticus (SRSE) during 2011-2017 at the Chinese University of Hong Kong, where Dr. Yau practices. Seven children died during their index hospital admission, with a median time to death of 8 days. Two more died within several years post discharge.
Morbidity was substantial: At follow-up 1 year after the index episode of SRSE, two patients had a Glasgow Outcome Scale (GOS) score of 3, indicative of severe disability; three patients had moderate disability, with a GOS of 4; and two patients were in a vegetative state, with a GOS of 2, both of whom subsequently died of aspiration pneumonia. Only 1 of the 15 patients had a good recovery. Through 8 years of follow-up, all six survivors had epilepsy. Common nonneurologic deficits included a predisposition to a variety of infections.
By way of background, Dr. Yau noted that convulsive status epilepticus is the most common neurologic emergency in children, with an incidence of about 20 episodes per 100,000. Of affected children, 10%-40% develop refractory status, with reported mortality rates of 16%-43%. SRSE is a term reserved for persistent or recurrent seizures 24 hours or more after onset of general anesthesia for management of refractory status.
The impetus for Dr. Yau’s study was the dearth of data on SRSE in children. The literature consists of a few case series totaling well under 100 patients.
The Hong Kong case series included 15 patients with SRSE who had a median age of 7.9 years, only 1 of whom had preexisting epilepsy, a case of epileptic encephalopathy with severe developmental delay. Of the 15, 12 were boys. The patients were placed on a median of four antiepileptic drugs. Those who survived to discharge spent a median of 17.8 days under general anesthesia and 42.5 days in the pediatric ICU.
The SRSE etiologies included febrile infection–related epilepsy syndrome in two cases, four serious infections, four cases of autoimmune etiology, two cases of epileptic encephalopathy, one patient with hypoxia caused by severe croup, and two of unknown origin despite intensive work-up.
The four in-hospital deaths caused by acute cerebral edema occurred a median 6.5 days after admission. There were also two deaths because of uncontrolled sepsis and one because of intraventricular bleeding secondary to thrombotic thrombocytopenic purpura thought to have occurred as a complication of interactions between the numerous prescribed medications. All six children with an infectious or unknown etiology died in hospital, whereas none of those with an autoimmune etiology, epileptic encephalopathy, or hypoxia did. Duration of anesthesia did not predict mortality.
Other investigators have reported that younger age is associated with higher mortality, but that was not true in the Hong Kong experience. Neither of the two children aged less than 3 years died during their index hospitalization. All 7 deaths occurred in the 13 children age 3 years or older.
When asked whether she thought SRSE or the underlying disorder was the bigger contributor to mortality, Dr. Yau replied that she believes the prolonged refractory seizures may have worsened cerebral edema in some patients and thereby have been the cause of death.
She reported having no financial conflicts regarding her study.
BANGKOK – presented by Maggie Lo Yee Yau, MD, at the International Epilepsy Congress.
“Death in these children usually occurred within the first few days after admission to the pediatric ICU,” she said at the congress sponsored by the International League Against Epilepsy.
The study included 15 consecutive patients aged between 1 month and 17 years treated for superrefractory status epilepticus (SRSE) during 2011-2017 at the Chinese University of Hong Kong, where Dr. Yau practices. Seven children died during their index hospital admission, with a median time to death of 8 days. Two more died within several years post discharge.
Morbidity was substantial: At follow-up 1 year after the index episode of SRSE, two patients had a Glasgow Outcome Scale (GOS) score of 3, indicative of severe disability; three patients had moderate disability, with a GOS of 4; and two patients were in a vegetative state, with a GOS of 2, both of whom subsequently died of aspiration pneumonia. Only 1 of the 15 patients had a good recovery. Through 8 years of follow-up, all six survivors had epilepsy. Common nonneurologic deficits included a predisposition to a variety of infections.
By way of background, Dr. Yau noted that convulsive status epilepticus is the most common neurologic emergency in children, with an incidence of about 20 episodes per 100,000. Of affected children, 10%-40% develop refractory status, with reported mortality rates of 16%-43%. SRSE is a term reserved for persistent or recurrent seizures 24 hours or more after onset of general anesthesia for management of refractory status.
The impetus for Dr. Yau’s study was the dearth of data on SRSE in children. The literature consists of a few case series totaling well under 100 patients.
The Hong Kong case series included 15 patients with SRSE who had a median age of 7.9 years, only 1 of whom had preexisting epilepsy, a case of epileptic encephalopathy with severe developmental delay. Of the 15, 12 were boys. The patients were placed on a median of four antiepileptic drugs. Those who survived to discharge spent a median of 17.8 days under general anesthesia and 42.5 days in the pediatric ICU.
The SRSE etiologies included febrile infection–related epilepsy syndrome in two cases, four serious infections, four cases of autoimmune etiology, two cases of epileptic encephalopathy, one patient with hypoxia caused by severe croup, and two of unknown origin despite intensive work-up.
The four in-hospital deaths caused by acute cerebral edema occurred a median 6.5 days after admission. There were also two deaths because of uncontrolled sepsis and one because of intraventricular bleeding secondary to thrombotic thrombocytopenic purpura thought to have occurred as a complication of interactions between the numerous prescribed medications. All six children with an infectious or unknown etiology died in hospital, whereas none of those with an autoimmune etiology, epileptic encephalopathy, or hypoxia did. Duration of anesthesia did not predict mortality.
Other investigators have reported that younger age is associated with higher mortality, but that was not true in the Hong Kong experience. Neither of the two children aged less than 3 years died during their index hospitalization. All 7 deaths occurred in the 13 children age 3 years or older.
When asked whether she thought SRSE or the underlying disorder was the bigger contributor to mortality, Dr. Yau replied that she believes the prolonged refractory seizures may have worsened cerebral edema in some patients and thereby have been the cause of death.
She reported having no financial conflicts regarding her study.
REPORTING FROM IEC 2019
Dupilumab found effective for adolescents with moderate to severe AD
AUSTIN, TEX. – according to results of a phase 3 study.

“Dupilumab works as effectively in adolescents as in adults,” Randy Prescilla, MD, one of the study authors, said in an interview at the annual meeting of the Society for Pediatric Dermatology. “It gives us promise that we could go into other age groups with the same optimism. We are enrolling patients in even younger age groups.”
The double-blind, placebo-controlled study analyzed the efficacy and safety of dupilumab monotherapy in patients between the ages of 12 and 17 years with moderate to severe atopic dermatitis (AD) inadequately controlled with topical therapies. In the United States, dupilumab is approved for those aged 12 years and older with moderate to severe disease inadequately controlled by topical prescription treatments or when those therapies are not advisable.
For the 16-week study, Dr. Prescilla, global medical affairs director of pediatric dermatology for Sanofi Genzyme, and colleagues randomized 251 patients to one of three groups: dupilumab every 2 weeks (200 mg if baseline weight was less than 60 kg; 300 mg if that weight was 60 kg or more); 300 mg dupilumab every 4 weeks; or placebo every 2 weeks.
At week 16, a significantly higher proportion of patients in the two drug treatment groups had Investigator’s Global Assessment scores of 0/1, compared with those in the placebo group (24.4%, 17.9%, and 2.4%) as well as a significantly higher percentage of patients who achieved at least a 75% improvement in the Eczema Area and Severity Index (EASI-75) score (41.5%, 38.1%, and 8.2%).
In addition, patients in the two drug treatment groups experienced improved percent change in least square-means on the EASI from baseline to week 16, compared with those in the placebo group (–65.9%, –64.8%, and –23.6%), the Peak Pruritus Numerical Rating Scale (–47.9%, –45.5%, and –19.0%), body surface area affected by AD (–30.1%, –33.4%, and –11.7%), and in the SCORing AD clinical tool (P less than .001 for all comparisons).
Between baseline and week 16, scores on the Children’s Dermatology Life Quality Index and Patient-Oriented Eczema Measure improved significantly in the two dupilumab groups, compared with the placebo group. The rate of skin infection was higher in the placebo group (20%), compared with 11% in the group that received dupilumab every 2 weeks and 13.3% in the group receiving the drug every 4 weeks.
Conjunctivitis occurred more frequently with dupilumab treatment (9.8% in the every-2-weeks dupilumab group, 10.8% in the every-4-weeks dupilumab group, and 4.7% in the placebo group) as did injection site reactions (8.5%, 6.0%, and 3.5%). Two adverse events, one of which was serious, occurred in the placebo group.
Dr. Prescilla acknowledged certain limitations of the study, including its small sample size and the fact that it was limited to 16 weeks. “However, smaller sample size and duration are typical for this type of study and in line with the study design of the SOLO 1 and SOLO 2 studies in adults,” he said.
On Aug. 6, the European Commission extended the marketing authorization for dupilumab in the European Union to include adolescents 12-17 years of age with moderate to severe atopic dermatitis who are candidates for systemic therapy. On the same day, Sanofi Genzyme and Regeneron announced positive topline results in a phase 3 trial in children aged 6-11 years with severe AD.
The study’s principal investigator was Amy S. Paller, MD. The study was funded by Sanofi Genzyme and Regeneron. Dr. Prescilla is an employee of Sanofi Genzyme.
AUSTIN, TEX. – according to results of a phase 3 study.
“Dupilumab works as effectively in adolescents as in adults,” Randy Prescilla, MD, one of the study authors, said in an interview at the annual meeting of the Society for Pediatric Dermatology. “It gives us promise that we could go into other age groups with the same optimism. We are enrolling patients in even younger age groups.”
The double-blind, placebo-controlled study analyzed the efficacy and safety of dupilumab monotherapy in patients between the ages of 12 and 17 years with moderate to severe atopic dermatitis (AD) inadequately controlled with topical therapies. In the United States, dupilumab is approved for those aged 12 years and older with moderate to severe disease inadequately controlled by topical prescription treatments or when those therapies are not advisable.
For the 16-week study, Dr. Prescilla, global medical affairs director of pediatric dermatology for Sanofi Genzyme, and colleagues randomized 251 patients to one of three groups: dupilumab every 2 weeks (200 mg if baseline weight was less than 60 kg; 300 mg if that weight was 60 kg or more); 300 mg dupilumab every 4 weeks; or placebo every 2 weeks.
At week 16, a significantly higher proportion of patients in the two drug treatment groups had Investigator’s Global Assessment scores of 0/1, compared with those in the placebo group (24.4%, 17.9%, and 2.4%) as well as a significantly higher percentage of patients who achieved at least a 75% improvement in the Eczema Area and Severity Index (EASI-75) score (41.5%, 38.1%, and 8.2%).
In addition, patients in the two drug treatment groups experienced improved percent change in least square-means on the EASI from baseline to week 16, compared with those in the placebo group (–65.9%, –64.8%, and –23.6%), the Peak Pruritus Numerical Rating Scale (–47.9%, –45.5%, and –19.0%), body surface area affected by AD (–30.1%, –33.4%, and –11.7%), and in the SCORing AD clinical tool (P less than .001 for all comparisons).
Between baseline and week 16, scores on the Children’s Dermatology Life Quality Index and Patient-Oriented Eczema Measure improved significantly in the two dupilumab groups, compared with the placebo group. The rate of skin infection was higher in the placebo group (20%), compared with 11% in the group that received dupilumab every 2 weeks and 13.3% in the group receiving the drug every 4 weeks.
Conjunctivitis occurred more frequently with dupilumab treatment (9.8% in the every-2-weeks dupilumab group, 10.8% in the every-4-weeks dupilumab group, and 4.7% in the placebo group) as did injection site reactions (8.5%, 6.0%, and 3.5%). Two adverse events, one of which was serious, occurred in the placebo group.
Dr. Prescilla acknowledged certain limitations of the study, including its small sample size and the fact that it was limited to 16 weeks. “However, smaller sample size and duration are typical for this type of study and in line with the study design of the SOLO 1 and SOLO 2 studies in adults,” he said.
On Aug. 6, the European Commission extended the marketing authorization for dupilumab in the European Union to include adolescents 12-17 years of age with moderate to severe atopic dermatitis who are candidates for systemic therapy. On the same day, Sanofi Genzyme and Regeneron announced positive topline results in a phase 3 trial in children aged 6-11 years with severe AD.
The study’s principal investigator was Amy S. Paller, MD. The study was funded by Sanofi Genzyme and Regeneron. Dr. Prescilla is an employee of Sanofi Genzyme.
AUSTIN, TEX. – according to results of a phase 3 study.
“Dupilumab works as effectively in adolescents as in adults,” Randy Prescilla, MD, one of the study authors, said in an interview at the annual meeting of the Society for Pediatric Dermatology. “It gives us promise that we could go into other age groups with the same optimism. We are enrolling patients in even younger age groups.”
The double-blind, placebo-controlled study analyzed the efficacy and safety of dupilumab monotherapy in patients between the ages of 12 and 17 years with moderate to severe atopic dermatitis (AD) inadequately controlled with topical therapies. In the United States, dupilumab is approved for those aged 12 years and older with moderate to severe disease inadequately controlled by topical prescription treatments or when those therapies are not advisable.
For the 16-week study, Dr. Prescilla, global medical affairs director of pediatric dermatology for Sanofi Genzyme, and colleagues randomized 251 patients to one of three groups: dupilumab every 2 weeks (200 mg if baseline weight was less than 60 kg; 300 mg if that weight was 60 kg or more); 300 mg dupilumab every 4 weeks; or placebo every 2 weeks.
At week 16, a significantly higher proportion of patients in the two drug treatment groups had Investigator’s Global Assessment scores of 0/1, compared with those in the placebo group (24.4%, 17.9%, and 2.4%) as well as a significantly higher percentage of patients who achieved at least a 75% improvement in the Eczema Area and Severity Index (EASI-75) score (41.5%, 38.1%, and 8.2%).
In addition, patients in the two drug treatment groups experienced improved percent change in least square-means on the EASI from baseline to week 16, compared with those in the placebo group (–65.9%, –64.8%, and –23.6%), the Peak Pruritus Numerical Rating Scale (–47.9%, –45.5%, and –19.0%), body surface area affected by AD (–30.1%, –33.4%, and –11.7%), and in the SCORing AD clinical tool (P less than .001 for all comparisons).
Between baseline and week 16, scores on the Children’s Dermatology Life Quality Index and Patient-Oriented Eczema Measure improved significantly in the two dupilumab groups, compared with the placebo group. The rate of skin infection was higher in the placebo group (20%), compared with 11% in the group that received dupilumab every 2 weeks and 13.3% in the group receiving the drug every 4 weeks.
Conjunctivitis occurred more frequently with dupilumab treatment (9.8% in the every-2-weeks dupilumab group, 10.8% in the every-4-weeks dupilumab group, and 4.7% in the placebo group) as did injection site reactions (8.5%, 6.0%, and 3.5%). Two adverse events, one of which was serious, occurred in the placebo group.
Dr. Prescilla acknowledged certain limitations of the study, including its small sample size and the fact that it was limited to 16 weeks. “However, smaller sample size and duration are typical for this type of study and in line with the study design of the SOLO 1 and SOLO 2 studies in adults,” he said.
On Aug. 6, the European Commission extended the marketing authorization for dupilumab in the European Union to include adolescents 12-17 years of age with moderate to severe atopic dermatitis who are candidates for systemic therapy. On the same day, Sanofi Genzyme and Regeneron announced positive topline results in a phase 3 trial in children aged 6-11 years with severe AD.
The study’s principal investigator was Amy S. Paller, MD. The study was funded by Sanofi Genzyme and Regeneron. Dr. Prescilla is an employee of Sanofi Genzyme.
REPORTING FROM SPD 2019